IJCRR - 13(7), April, 2021
Pages: 172-184
Date of Publication: 12-Apr-2021
Print Article
Download XML Download PDF
Comparative Quality Analysis of Cefpodoxime Proxetil Branded Tablet Product with Available Indian Generic Products: Short-Term Accelerated Study
Author: Darshana R. Dumbhare, Fahimuddin S. Kazi, Debarshi Kar Mahapatra, Ujwala N. Mahajan
Category: Healthcare
Abstract:Introduction: Cefpodoxime is a safe, short course, effective, semi-synthetic, third-generation cephalosporin available for use as a prodrug known as Cefpodoxime proxetil and is recommended for treating sinusitis, otitis media, urinary tract infection, respira�tory tract infections (upper and lower), skin infections, and soft tissue infections. It is available both in the form of branded prod�ucts and generic products where both have been seen to have different quality, physicochemical, and stability characteristics. Objective: To explore the quality attributes of a branded cefpodoxime proxetil product and five different generic cefpodoxime proxetil products available in the Indian market. Methods: The present exploration involved investigating the quality attributes (Assay determination and Impurity testing), phys�icochemical test (Physical appearance of tablets, Packaging and labelling of tablets, Tablet diameter and thickness, Weight variation test, Friability Test, Hardness test, Disintegration test, and In vitro dissolution study), and accelerated stability study (1 month, 3 months, and 6 months under temperature 40\?C \? 2\?C and humidity 75% \? 5% RH) of a branded cefpodoxime proxetil product and five different generic cefpodoxime proxetil products available in the Indian market as per the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines (Q2 and Q3) and the United States Pharmacopeia (USP) guidelines. A comparison between the branded product and the generic products has been made in terms of the quality attributes. Results: All the generics and the branded cefpodoxime proxetil tablets were found to be within the acceptable range. Conclusion: The study will open new doors of analysis of pharmaceutical quality assurance and provide avenues for both branded products and generic products in maintaining the quality attributes regularly
Keywords: Cefpodoxime proxetil, Branded, Generic, Accelerated Stability Study, Impurity, Degradation
Full Text:
INTRODUCTION
The concept of quality assessment is an absolute system to create and follow the practices and strategies for providing the most consistent laboratory outcomes and to lessen the errors involved in the pre-analytical, analytical, and post-analytical stages.1 Among the two foremost elements, Quality Assessment is one of the universal quality management systems. Performance Improvement is one of the headways that have activities related to the activity of the other processes.2 Quality assessment service in context to growing organizational stages principally highlighting the data quality and the detection, particularly contained by the monetary sectors, that data correctness and veracity are critical to execution with lawmaking and regulatory approvals.3
Cefpodoxime is a safe, short course, effective, semi-synthetic, third-generation cephalosporin available for use as a prodrug known as Cefpodoxime proxetil.4 It is absorbed readily from the human gastrointestinal tract. It is an active drug given in various regimens for diverse infections caused by Gram-positive cocci such as streptococci, staphylococci, penicillinase-producing strains as well as Gram-negative bacteria such as Meningococci, Hemophilus, Gonococci, Klebsiella, Moraxella, E. coli, etc.5 It is highly recommended for treating sinusitis, otitis media, urinary tract infection, respiratory tract infections (upper and lower), skin infections, and soft tissue infections.6 The drug can be recommended as a parenteral cephalosporin with a maximum dose of 8–10 mg/kg/d.7 It is majorly excreted by the kidneys in an unchanged form. In renal compromised patients, dose adjustments are required.8 It attains sufficient stages beyond the minimum inhibitory concentration (MIC) in several body fluids, therefore, the doses need to be adjusted (single or double doses).9
A branded medication is an innovative creation that has been created by an innovator organization. When an innovator develops a new medication, the developed novel drug product must endure and pass recommended quality tests and evaluate to make sure its effectiveness in remedial situations to treat and safely administer for human use.10 As innovator organizations spend a substantial amount of resources to formulate a novel drug product, they are provided with the exclusive right to produce and market the medications for a given duration.11 When an organization is provided exclusive rights to produce and market, the medicament is credited to patent(s). For a given duration of the patent award to the original company, no other organizations have the right to manufacture the same drug.12 For this motive, a branded medicine is well known and that particular medication is generally very much trusted.
A generic medicine may be defined as a drug product that corresponds to a meticulous reference listed drug product (RLD) or drug brand in the context of strength, type of dosage form, route of administration, performance characteristics, quality, and its application, which may be distinguished as a pharmaceutical equivalent, bioequivalent, and chemical equivalent to RLD.13 A generic medicine, is in reality, a replica of the innovative recognized drug product.14 Once the lawful exclusive rights for the original product have been exhausted over time, the organizations that innovated the product are devoid of the elite certified right to create and share out the medicine.15 Other organizations will produce the same adaptation of the identical medication with the equivalent excellence characteristics and can be put up for trade in the marketplace at a much reasonable cost.16
The present exploration involved investigating the quality attributes (Assay determination and Impurity testing), physicochemical test (Physical appearance of tablets, Packaging and labelling of tablets, Tablet diameter and thickness, Weight variation test, Friability Test, Hardness test, Disintegration test, and In vitro dissolution study), and accelerated stability study (1 month, 3 months, and 6 months under temperature 40°C ± 2°C and humidity 75% ± 5% RH) of a branded cefpodoxime proxetil product and five different generic cefpodoxime proxetil products available in the Indian market as per the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines (Q2 and Q3) and the United States Pharmacopeia (USP) guidelines. A comparison between the branded product and the generic products has been made in terms of the quality attributes.
MATERIALS AND METHODS
Instruments
The UV-Vis spectroscopic analysis was performed using the double-beam Shimadzu® Ultraviolet-Visible Spectrophotometer (Model: UV-1800, Japan) which was connected with a computer desktop system. The system has a spectral bandwidth of 1 nm with wavelength accuracy of ±0.3 nm and also comprises a pair of matched quartz cells having a 10 mm path length. All weighing of chemicals were carried out using Wensar® high precision electronic balance (Model: PGB100, USA). The sonication was done using the Transonic Digital S (Sonicator), USA. The hardness testing was achieved by using a Monsanto hardness tester (Model: Campbell, USA). Electrolab® disintegration tester USP (Model: ED2L) was employed for studying the disintegration of the tablets. Electrolab® dissolution tester (Model: TDT-08L) was utilized for studying the dissolution of the tablets. Electrolab® Roche Friabilator (Model: EF2) was used for studying the friability of the tablets. The HPLC study was carried out on a Waters® 2695 system with PDA detector 2996 on a reverse-phase Denali C18 column (150 mm × 4.6 mm dimension, 5 μm particle size). The system was equipped with EMPOWERS v.2 software comprising of a 20-μl loop manual rheodyne injector.
Chemicals
HPLC grade methanol, acetonitrile, glacial acetic acid, and water, as well as analytical grade glycine, sodium chloride, and ammonium acetate, were procured from Himedia Ltd., Mumbai, India. The cefpodoxime proxetil branded product and the five generic cefpodoxime proxetil products were purchased from various Pharmacies across the city limit of Nagpur, Maharashtra, India.
Spectral analysis of cefpodoxime proxetil
Preparation of stock solution
Accurately weighed 25 mg of cefpodoxime proxetil was transferred in a 25 mL volumetric flask and further dissolved in methanol. The final volume of this stock solution (A) was made up to 25 mL with methanol to make 1000 µg/mL concentration.
Preparation of dissolution medium pH 3.0
In a 1000 mL volumetric flask, 54.5 g of glycine and 42.6 g of sodium chloride were dissolved in 500 mL of water and 14.2 mL of hydrochloric acid was added with swirling. The content was allowed to cool, further diluted with water, and mixed well. 50 mL of the stock solution was transferred to a volumetric flask and diluted with 900 mL of water to obtain a solution. A pH of 3.0 ± 0.1 of the stock solution was adjusted with 10 N NaOH.
Determination of λmax for dissolution
From the above stock solution (A), 10 ml of the solution was taken and further diluted to 100 mL with dissolution medium pH 3.0 to make 100 µg/mL concentration (referred to as stock solution (B)). From the above solution, 25 mL content was taken and again diluted to 100 mL with dissolution medium pH 3.0 to make 25 µg/mL concentration (referred to as stock solution (C)). This solution was scanned in the range of 400 nm to 200 nm using a blank and the λmax was determined.
Preparation of standard curve of cefpodoxime proxetil
The dilutions were prepared from the stock solution (C) where the UV absorbance of 5 µg/mL, 10 µg/mL, 15 µg/mL, 20 µg/mL, and 25 µg/mL solutions were measured on UV-visible spectroscopy at 261 nm for the preparation of standard curve of cefpodoxime proxetil.
Evaluation parameters for marketed branded and generic products
The test methods given under the USP monograph for cefpodoxime proxetil tablets were employed for the evaluation of all products.
Tablet description
The color, shape, and size were examined by visual observation.
Tablet Diameter and Thickness
The diameter and thickness of tablets are important for determining the uniformity of tablet size. Thickness and diameter were measured using Screw Guage Micrometer, which permits accurate measurements and provides information on variation between tablets.17
Weight variation
As per the USP, the weight variation test was performed by weighing 20 tablets individually, calculating the average weight, and comparing the individual tablet weight with that of the average. According to the specification outlined in USP, the test for the uniformity of weight for drug products; 130-324 mg is ±7.5% and >324 mg is ±5% of the average pass. Then, the tolerance limit for weight variation was estimated.18
Tablet hardness
The tablets require a certain amount of strength and resistance to friability and also to withstand mechanical shocks during handling in manufacture, packaging, and shipping. It is the property of a tablet that is measured to assess its resistance to permanent deformation. The tablet hardness tester device (Monsanto tester) was used to test tablet hardness. The hardness of each branded tablet and generic tablets were measured in unit kg/cm2. Each sample was analyzed in a triplicate manner.19
Disintegration study
Disintegration is the breakdown of the tablets into smaller particles or granules when it comes in contact with a solution. The time taken by a tablet to disintegrate was measured in a USP disintegration apparatus. For determining the disintegration time, one tablet was placed in each tube and the basket rack was positioned in 1 L simulated gastric fluid without enzymes at pH 1.2 under a temperature of 37°C ± 1°C. The frequency of cycles per minute was 28 to 32.20
Dissolution study
The release rate of cefpodoxime proxetil film-coated tablets was determined by using USP dissolution testing apparatus-II (Paddle type). The dissolution test was performed using 900 mL (0.1 N HCl) volume at a temperature of 37°C ±0.5°C with 100 rpm stirring. An aliquot (10 mL) of the solution was collected from the dissolution vessel, filtered through a 0.45 µm Whatman filter paper, and diluted 2 mL of the filtered content with a dissolution medium at time intervals 10 min, 20 min, and 30 min. Each aliquot was replaced with a fresh dissolution medium to maintain the sink conditions. The absorbance of diluted solutions was measured at 261 nm.21
Accelerated stability studies
The branded cefpodoxime proxetil product and generic cefpodoxime proxetil products were placed inside a polyvinyl chloride (PVC) container and aluminium foil was wrapped over it. The stability studies were performed under the accelerated conditions of temperature (40°C ± 2°C) and moisture (75% ± 5% RH) for the duration of 1 month, 3 months, and 6 months.22
Assay preparation
Preparation of mobile solution
A filtered and degassed mixture of 0.02 M ammonium acetate and acetonitrile was prepared in the ratio of 6:4.
Preparation of diluent
A filtered and degassed mixture of water and acetonitrile was prepared in the ratio of 6:4.
Standard preparation
25 mg of cefpodoxime proxetil was weighed accurately and transferred in a 50 ml of volumetric flask. The content was dissolved in 5 mL of methanol, further diluted with the diluent to the desired volume and mixed. 5 mL of the above solution was transferred to 100 mL of the volumetric flask, further diluted with the diluent to the desired volume, mixed, and passed through a filter having 0.45-μm porosity.
Assay procedure
Finely powdered 20 tablets were accurately weighed and a portion of the powder equivalent to 50 mg of cefpodoxime proxetil was transferred to a 100 mL volumetric flask. The content was dissolved in 10 mL of methanol, further diluted with the diluent to the desired volume and mixed. 5 mL of the above solution was transferred to 100 mL of the volumetric flask, further diluted with the diluent to the desired volume, mixed, and passed through a filter having 0.45-μm porosity.
Chromatographic system suitability
The liquid chromatography system was equipped with a 235 nm detector having a 250 mm × 4.6 mm dimension packing column. The flow rate was 2 mL/min at ambient temperature (30°C). The standard and the test preparations were chromatographed and the peak responses were recorded. About 0.9 for S-epimer cefpodoxime proxetil and 1.0 for R-epimer cefpodoxime proxetil are the relative retention periods. The resolution, R, is not less than 2.5 between the S-epimer cefpodoxime proxetil and the R-epimer cefpodoxime proxetil. No more than 1.5 is the cefpodoxime proxetil R-epimer tailing factor and no more than 1.0 percent is the relative standard deviation measured for replication injections from the cefpodoxime proxetil S-epimer and cefpodoxime proxetil R-epimer regions. Not more than 1.0% for any peak at relative retention times is desirable. Not more than 0.5% of any other individual impurity and not more than 6.0% of total impurities is desired. Not more than 3.0% of any peak at a relative retention time and individual peaks having relative retention times not higher than 2.0 is desired.
The percentage of each impurity in the portion of tablets was calculated by the formula:
% purity = 2000 (CP/W) (rU/rS)
C is the concentration (in mg per mL) of cefpodoxime proxetil in the standard preparation; P is the designated potency (in μg per mg) of cefpodoxim in USP cefpodoxime proxetil; W is the weight (in mg) of cefpodoxime proxetil taken to prepare the assay preparation; rU and rS are the peak response numbers obtained from the preparation of the assay and the standard preparation of cefpodoxime proxetil S-epimer and cefpodoxime proxetil R-epimer, respectively.
Impurity profiling
Preparation of Solution-A
Solution A was prepared by taking filtered and degassed 0.02 M ammonium acetate.
Preparation of Solution-B
Solution A was prepared by taking filtered and degassed acetonitrile.
Preparation of the mobile phase
The mobile phase of the chromatographic system involved the utilization of variable mixtures of solution-A and solution-B.
Preparation of diluent
A filtered and degassed mixture of water and acetonitrile was prepared in the ratio of 2:1.
System suitability solution
A quantity of cefpodoxime proxetil was dissolved in the diluent to obtain a solution containing 10 µg/mL. A volume of methanol not exceeding 10% of the total volume in the final solution may be used to facilitate dissolution.
Standard solution
50 mg of cefpodoxime proxetil was accurately weighed and transferred to a 50 ml of volumetric flask. The content was dissolved in 5 mL of methanol, using sonication if necessary, diluted with diluent to the desired volume, and mixed well. The solution was injected promptly and analyzed within 24 hours when stored at 8ºC temperature.
Test solution
50 mg of cefpodoxime proxetil was accurately weighed and transferred to a 50 ml of volumetric flask. The content was dissolved in 10 mL of methanol, using sonication if necessary, diluted with diluent to the desired volume, and mixed well. The solution was injected promptly and analyzed within 24 hours when stored at 8ºC temperature.
Chromatographic system
The liquid chromatography system was equipped with a 261 nm detector having 250 mm × 4.6 mm dimension packing column (5 μm particle size). The flow rate was 2 mL/min. The standard and the test preparations were chromatographed and the peak responses were recorded according to the following gradient procedure: 0 min [equilibration (10 min), solution A (90%), solution B (10%)]; 0-10 min [linear gradient, solution A (90%→68%), solution B (10%→32%)]; 10-40 min [isocratic, solution A (68%), solution B (32%)]; 40-80 min [linear gradient, solution A (68%→50%), solution B (32%→50%)]; 80-85 min [isocratic, solution A (50%), solution B (50%)]; 85-90 [linear gradient, solution A (50%→25%), solution B (50%→75%)]; 90-95 [isocratic, solution A (25%), solution B (75%)]; 95-100 [linear gradient, solution A (25%→90%), solution B (75%→10%)]. The tailing factor and the relative standard deviation for replicate injection should not more than 2.0.
Assay Procedure
20 μL of test solution was injected into the chromatography system and the chromatogram was recorded. All the measurements were done by the peak areas. The percentage of each impurity in the portion of cefpodoxime proxetil was obtained from the formula:
% Impurity = 100 (ri / rs)
Where ri is the peak area for each impurity and rs is the sum of the areas of all the peaks.
RESULTS AND DISCUSSION
Spectral analysis of cefpodoxime proxetil
lmax determination
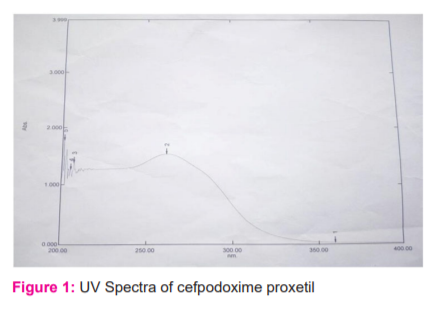
The prominent wavelengths at 261.40 nm, 213.20 nm, and 208.80 nm were predominantly seen (Figure 1). An absorption maximum was found to be at 261 nm. Hence, 261 nm was selected as the lmax for further studies.
Prepared standard curve
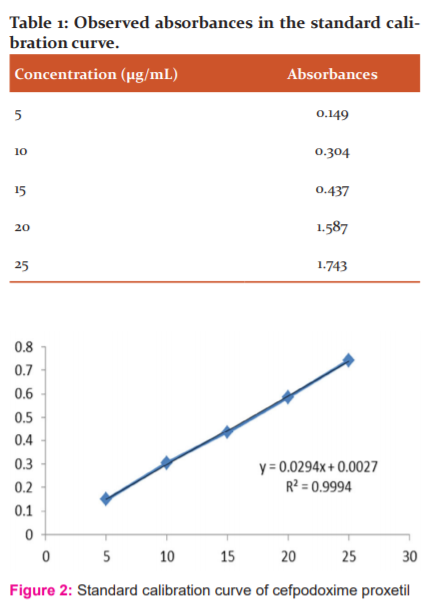
Cefpodoxime proxetil showed maximum absorption at wavelength 261 nm in glycine medium. The calibration curve was prepared by taking the UV absorption of solutions at 5-25 µg/mL concentration (Table 1). The details of calibration curve include y = 0.0294x + 0.0027 with R2 = 0.9994 (Figure 2).
Evaluation parameters for marketed branded and generic products
Tablet description
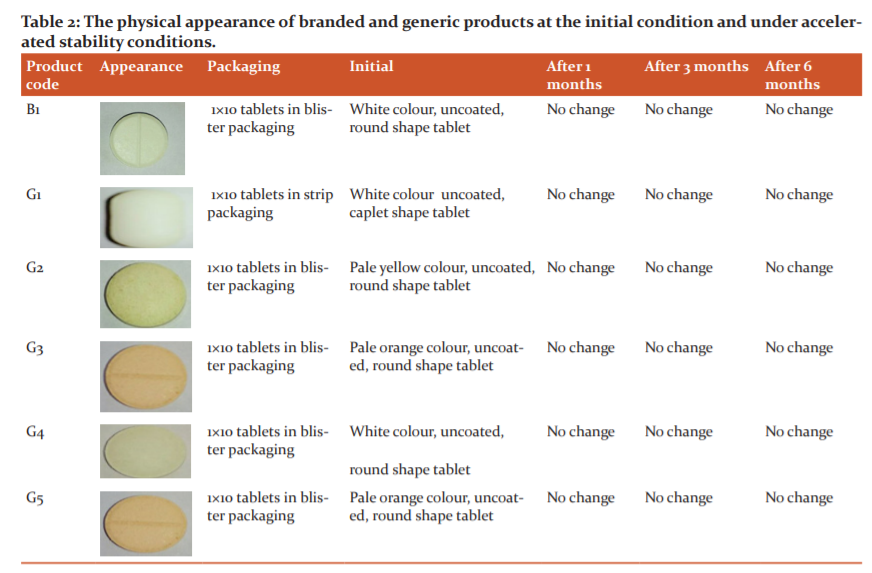
The colour, shape, and size were examined by visual observation where the factory manufactured cefpodoxime proxetil products (1×10 tablets in blister packaging) had the same attributes as per pharmacopoeia recommendation and the manufacturer’s claim (Table 2). The visual appearance of the product is an important factor for patient compliance. The branded product was a white colour, uncoated, round shape tablet whereas the generic products were white color uncoated, caplet shape tablet (G1 product), pale yellow color, uncoated, round shape tablet (G2 product), White color, uncoated, round shape tablet (G4 product), and pale orange color, uncoated, round shape tablet (G3 and G5 products). After performing the accelerated stability studies, no changes were observed. The physical appearances and the physical stability of the products were found to be very uniform.
Tablet Diameter and Thickness
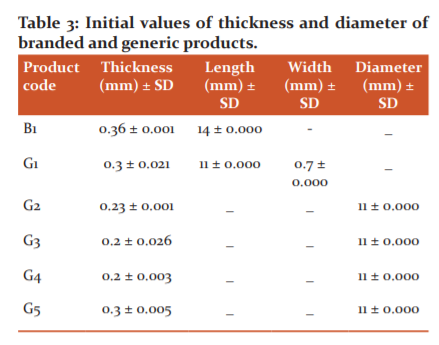
All the generics and the branded cefpodoxime proxetil tablets were found to be within the acceptable range of thickness (0.2-0.36 mm), length (11-14 mm), width (0.7 mm), and diameter (11 mm). The thickness and the diameter uniformity of the tablets are a necessary factor for the consumer requirements and also important for better packaging. No changes occurred in branded and generic products after the stability study (Table 3).
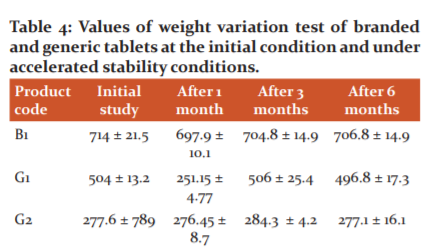
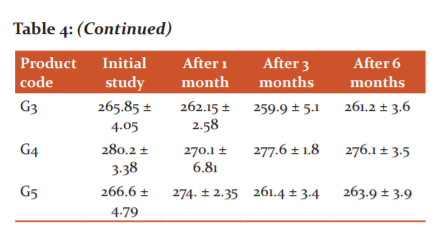
Weight variation
The average weight of 20 tablets was calculated and the individual tablet weight was compared to the average weight. The weight variation test is required to assure that the drug content in each unit dose is distributed in a narrow range around the label strength. If the drug substance forms the greater part of the oral solid dosage form, the weight variation reflects variation in the content of the active ingredient. The results indicate that five generics (G1-G5) products and one branded (B1) product possess acceptable uniformity of weight as per the USP limit. After the stability shelf period, there were no prominent changes observed (Table 4).
Tablet hardness
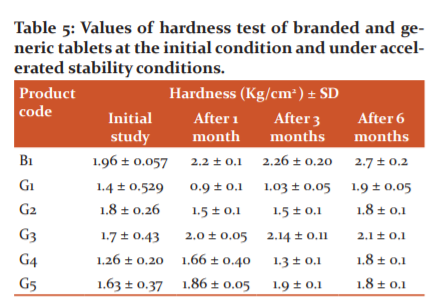
The hardness test showed the ability of tablets to withstand pressure or stress during handling, packaging, and transportation. The results indicated that the branded tablet product B1 had the highest strength; i.e. 1.96 kg/cm2 as compared to all the five generic products (G1-G5). The generic products G2 and G5 retained the hardness across the 6 months accelerated stability period while branded tablet product B1 as well as generics G1, G3, and G4 showed an increase in the hardness value after 6 months of the accelerated stability period. The reason for the augmentation of hardness value (breaking strength) may be due to the specific excipients used, moisture absorption, and temperature. Generic products G1, G2, G4, and G5 showed a hardness value of less than 2 kg/cm2, after accelerated stability studies which are not a good sign of compressibility and therefore the batches failed. Only branded tablet product B1 and generic product G3 showed a good sign for tablets dosage form (Table 5).
Friability study
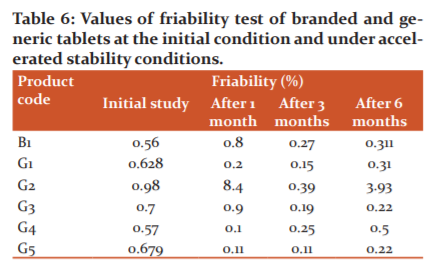
6 tablets of each product were used for the friability test. The branded product B1 and generic tablet products (G1-G5) demonstrated friability of not more than 1% in both initial study and for 6 months accelerated stability study. The friability values were observed to be decreased over time which may be due to retaining product hardness owing to moisture absorption and temperature. All these products passed the friability test (Table 6).
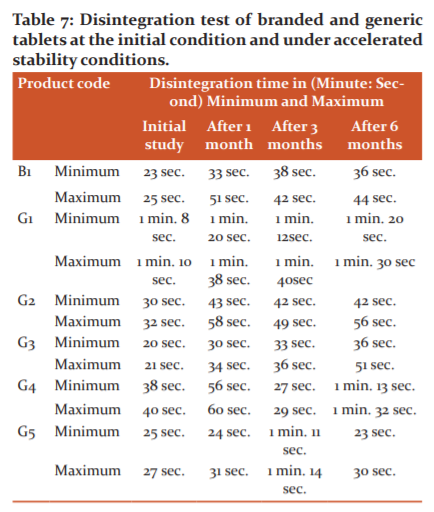
Disintegration study
The disintegration test is the most important step in the release of drugs from the immediate release dosage forms. The rate of disintegration is directly proportional to the rate of dissolution. The rate of disintegration is straightly influenced by the rate of influx of water into the tablets and also depends on the porosity of the tablets. The above results indicated that the branded product B1 and three generic products G2, G3, and G5 passed the disintegration test at initial levels but branded product B1 and two generic products G2 and G3 failed after the accelerated stability period according to the USP guidelines (Table 7). It was observed that the G1 generic product displayed out of limit at both initial and accelerated stability levels. Generic products G4 and G5 presented variable results after the 3rd month and 6th month of accelerated stability period which may be due to tablet to tablet variation.
Dissolution study
The dissolution test of the drug from a solid dosage form is important for drug bioavailability. In the dissolution study, 6 tablets of one branded product B1 and generic products (G1-G5) were tested for the % drug release, according to the method described for the cefpodoxime proxetil tablet in the USP monograph. It is stated in the monograph that the amount of cefpodoxime proxetil released within 30 minutes should not less than 70% of the stated amount.
The in vitro dissolution rate is used to simulate the bioequivalency of different formulation of generics (cefpodoxime proxetil tablet) concerning the branded product. Initial dissolution study was performed and the results suggested that the branded product B1, generic products G1 and G3 passed the test while generic products G4 and G5 were at borderline; i.e. ~71% while the generic batch G2 failed in the dissolution testing. The reason may be due to the altered physical characteristics and content non-uniformity.
The 1st month accelerated stability samples were analyzed for the dissolution test. Branded product B1 passes with best results; i.e. 86.89% after 30 min. The generic products G1 and G3 were observed to release the drugs at 73.26% and 72.68%, respectively (Table 8). At the same time, generic product G5 passes the critical lower limit of drug release; i.e. 71.8%. However, generic products G2 (59.74%) and G4 (58.71%) failed to show desired dissolution results. A great effect of temperature and humidity may be the plausible reason. Generic product GS4 passed the initial study with a close limit but after 1-month stability, a decrease in the drug dissolution was perceived due to poor production batch.
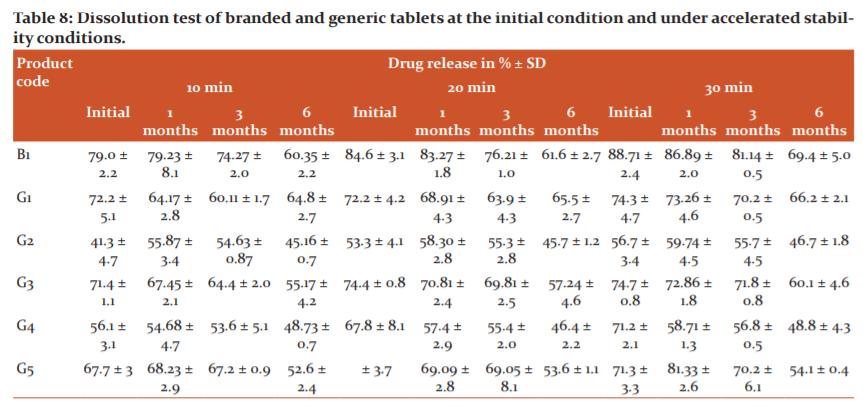
The 3rd month accelerated stability results presented that the branded product B1 passed the dissolution test while the generic products G1, G3, and G5 demonstrated the % drug release close to the limit; i.e. 70%. In contrast to it, two generic products G2 and G4 failed to meet the desired limit. According to Biopharmaceutics Classification System (BCS), cefpodoxime proxetil comes under Class-II (high permeability, low solubility) where it is expected that the dosage form should release >70% drug and as per specification (30 min). There was a decrease in the drug release for the branded product B1 and the generic products G1, G3, and G5, they closely met the required limit. The generic products G2 and G4 failed.
The 6th month accelerated stability results concluded that the branded product B1, as well as generic products (GS1-GS5), failed the dissolution study. Only the branded product B1 complied with the dissolution test until 3rd-month stability while generic products G1, G3, and G5 passed at a boundary level till the 3rd month stability period. Generics G2 and G4 were regarded as failed products during the initial study to 6 months accelerated stability study (40°C ± 2°C and 75°C ± 5% RH).
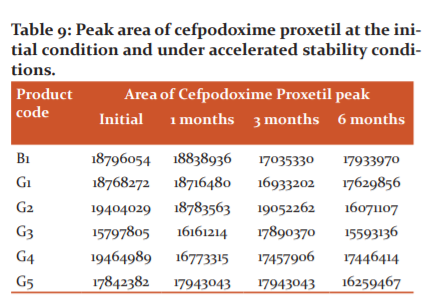
Assay of products
All the five generics products and one branded product were assayed for the drug content according to the isocratic mode based HPLC method (0.02 M ammonium acetate and acetonitrile in the ratio of 6:4) outlined in the individual drug monographs of the USP (acceptance limit: 90-110%). The content of cefpodoxime proxetil equivalent to cefpodoxime was calculated from the peak areas of the chromatograms of test and reference standard solutions (Table 9).
References:
-
Brook RH, Williams KN, Avery AD. Quality assurance today and tomorrow: forecast for the future. Annal Inter Med 1976;85(6):809-17.
-
Kessner DM. Quality assessment and assurance: early signs of cognitive dissonance. New Eng J Med 1978;298(7):381-6.
-
Farris KB, Kirking DM. Assessing the quality of pharmaceutical care II. Application of concepts of quality assessment from medical care. Annal Pharmacother 1993;27(2):215-23.
-
Frampton JE, Brogden RN, Langtry HD, Buckley MM. Cefpodoxime proxetil. Drugs. 1992;44(5):889-917.
-
Chugh K, Agrawal S. Cefpodoxime: pharmacokinetics and therapeutic uses. Indian J Pediatr 2003;70(3):227-31.
-
Todd WM. Cefpodoxime proxetil: a comprehensive review. Int J Antimicrob Agent 1994;4(1):37-62.
-
Fulton B, Perry CM. Cefpodoxime proxetil. Paediatr Drugs 2001;3(2):137-58.
-
Borin MT. A review of the pharmacokinetics of cefpodoxime proxetil. Drugs 1991;42(3):13-21.
-
Chocas EC, Paap CM, Godley PJ. Cefpodoxime proxetil: a new, broad-spectrum, oral cephalosporin. Annal Pharmacother 1993;27(11):1369-77.
-
Ahire K, Shukla MA, Gattani MA, Singh VI, Singh ME. A survey-based study in the current scenario of generic and branded medicines. Int J Pharm Pharm Sci 2013;5(3):705-11.
-
Bera A, Mukherjee A. The importance of generic drugs in India. Int J Pharm Chem Biol Sci 2012;2(4):575-87.
-
Dadhich A, Upadhyaya M. A review: exploring branded generic drugs by Indian pharmaceutical multinational companies as a new prospect. Pharmacophore 2011;2(6):271-5.
-
Greene JA. Generic: The unbranding of modern medicine. JHU Press; 2014.
-
Van der Merwe SE, Bredenkamp J. Originator and generic medicine: pricing and market share. Int J Pharm Healthc Mark 2013;7(2):104-19.
-
Dylst P, Vulto A, Godman B, Simoens S. Generic medicines: solutions for a sustainable drug market? Appl Health Econ Health Policy 2013;11(5):437-43.
-
Simoens S. International comparison of generic medicine prices. Curr Med Res Opin 2007;23(11):2647-54.
-
Mahajan NM, Pardeshi A, Mahapatra DK, Darode A, Dumore NG. Hypromellose and Carbomer induce bioadhesion of Acyclovir tablet to vaginal mucosa. Indo Am J Pharm Res 2017;7(12):1108-18.
-
Mahajan NM, Wadhwane P, Mahapatra DK. Rational designing of sustained-release matrix formulation of etodolac employing hypromellose, carbomer, eudragit and povidone. Int J Pharm Pharm Sci 2017;9(12):92-7.
-
Patil MD, Mahapatra DK, Dangre PV. Formulation and in-vitro evaluation of once-daily sustained release matrix tablet of nifedipine using rate retardant polymers. Inventi Impact Pharm Tech 2016;4:190-6.
-
Gangane PS, Kadam MM, Mahapatra DK, Mahajan NM, Mahajan UN. Design and Formulating Gliclazide Solid Dispersion Immediate Release Layer and Metformin Sustained Release Layer in Bilayer Tablet for the effective Postprandial Management of Diabetes Mellitus. Int J Pharm Sci Res 2018;9(9):3743-56.
-
Gangane PS, Ghughuskar SH, Mahapatra DK, Mahajan NM. Evaluating the Role of Celosia argentea Powder and Fenugreek Seed Mucilage as Natural Super-Disintegrating Agents in Gliclazide Fast Disintegrating Tablets. Int J Curr Res Rev 2020;12(17):101-8.
-
Godbole MD, Mahapatra DK, Khode PD. Fabrication and characterization of edible jelly formulation of stevioside: a nutraceutical or OTC aid for diabetic patients. Inventi Rapid: Nutraceut 2017;2:1-9.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





