IJCRR - 8(13), July, 2016
Pages: 42-49
Date of Publication: 12-Jul-2016
Print Article
Download XML Download PDF
SYNTHESIS AND APPLICATIONS OF SUGAR FLUORINATED NUCLEOSIDES
Author: Mohamed Ibrahim Elzagheid
Category: Healthcare
Abstract:In this review, different methods that have been used for the synthesis of 1'-, 2'-, 3'-, 4'- and 5'-sugar fluorinated nucleosides and their analogous are presented and different fluorinating agents are listed. Highlighted examples of the sugar fluorinated nucleosides that make a great impact on chemistry, biochemistry, and drug discovery are also elaborated. This review has shown that introduction of a fluorine atom in different positions within the sugar structure of the nucleoside improves their reactivity and properties.
Keywords: Nucleosides synthesis and applications, Sugar fluorinated nucleosides, Fluorinating agents
Full Text:
INTRODUCTION
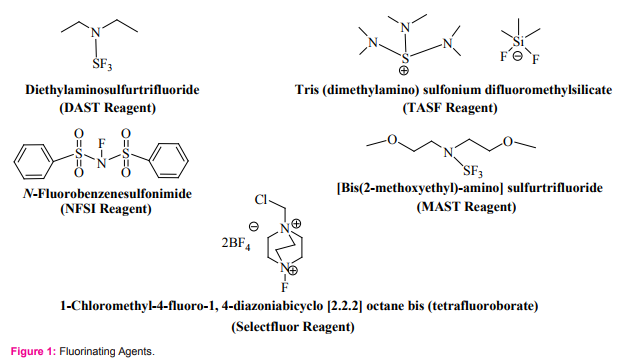
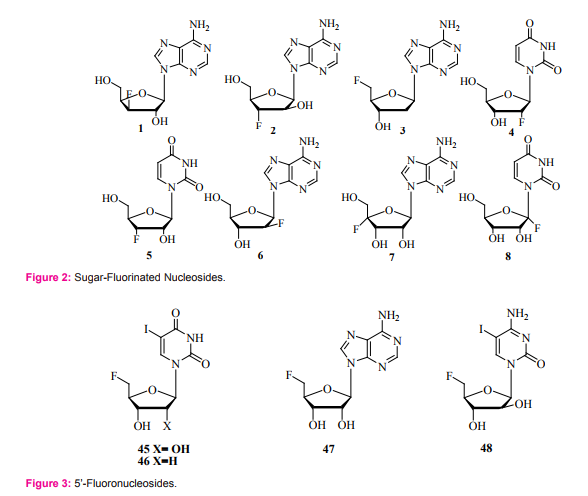
Fluorinated nucleosides attracted many medicinal chemists because of their promising medicinal applications. They play a major role in interrupting the replication of cancer cells or viruses [1-3].A great intention was given by chemists to the introduction of fluoro group into sugars and nucleosides and different methods for the formation of fluorine carbon bond have been developed [4-7]. It is believed that the inherited biological activity of the fluorine incorporated nucleosides may be due to the electronegativity of fluorine and the strength of the carbon-fluorine bond. Strong electronic character of fluorine can alter the electronic properties of the nucleosides and induce interesting biophysical properties to the sugar-fluorinated nucleosides [8]. They can also be used as building blocks in oligonucleotides [9]. The synthesis of fluorinated nucleosides (by introduction of fluorine atom at 1’, 2’, 3’, 4’, and 5’ positions) can be achieved, by different fluorinating agents (Figure 1), through selective fluorination of nucleosides or by glycosylation of nucleic acids bases (nucleobases) with fluoro-sugar derivatives [10-14]. There are six reported methods for the synthesis of sugarfluorinated nucleosides (Figure 2). These are:
1. Epoxide cleavage by fluoride ions: This reaction has been used for the synthesis of 9-(3-deoxy-3-fluoro-β- D-xylofuranosyl) adenine 1 [15], and 9-(3-deoxy-3- fluoro-β-D-arabinofuranosyl) adenine 2 [16].
2. Displacement of sulfonyloxy group by fluoride ions: This reaction has been used for the synthesis of 5’-fluronucleosides because displacement of 5’-sulfonates with fluorine is more easily achieved than the same reaction with 2’- or 3’-sulfonates. A good example is the synthesis of 9-(3, 5-dideoxy-5-fluoro -β-D-ribofuranosyl) adenine 3 [17].
3. Fluorine anhydro ring opening: This reaction has also been used for the introduction of fluorine in 2’- and 3’-postitions: A good example is the synthesis of 1-(2-deoxy-2-fluoro-β-D-ribofuranosyl) uracil 4 [18] and 1-(3-deoxy-3-fluoro-β-D-ribofuranosyl) uracil 5 [18].
4. One step exchange of hydroxyl group by fluorine: This has been achieved by treating desired sugar with diethylaminosulfurtrifluoride, DAST, or [bis (2-methoxyethyl)-amino] sulfurtrifluoride, MAST, then coupling the brominated fluorosugar with the nucleobases [10]. A good example is the synthesis of 9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl) purine 6 [10].
5. Sequential photo-bromination and fluorination: In this method desired sugar was treated with N-bromosuccinimide, NBS, followed by fluorination with silver tetrafluoroborate (AgBF4 ) [19]. A good example here is the synthesis of 4’-fluoroadenosine 7.
6. Electrophilic fluorination of the nucleoside 1’-position: in this synthesis treatment of the 1’-lithium enolate, prepared from the 2’-ketoderivative, with N-fluorobenzenesulfonimide (NFSI) or 1-Chloromethyl-4-fluoro-1, 4-diazoniabicyclo [2.2.2] octane bis (tetrafluoroborate) (Selectfluor) , followed by reduction of the 2’-keto-moeity gave the 1’-fluorouridine derivatives [20, 21]. A good example is the synthesis of 1’-fluorouridine 8 (unprotected nucleosides was not successfully isolated because of the instability.
Although excellent reviews on the synthetic aspects of the fluorinated nucleosides have been published [12, 22],some of them were long and sometimes confusing. Our present review is short, clear and to the point. We tried here to put more emphasis on synthetic methodology and applications of the sugar fluorinated nucleosides.
DISCUSSION
1’- Fluorinated nucleosides
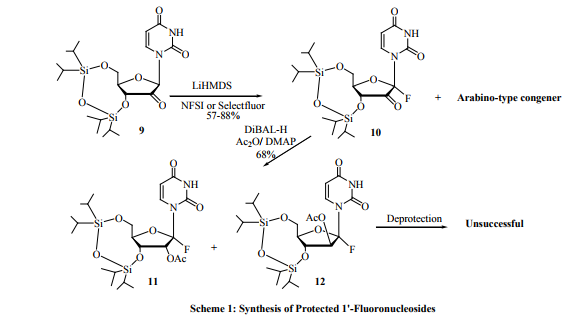
Almost all hydrogens attached to carbons have been substituted by fluorine atoms. However, substitution at 1’-carbon is rarely reported. This is may be due the feeling that 1’-nucleosides are difficult or unstable to synthesize. Recently the synthesis of protected 1’-fluoronucleosides (Scheme 1) was reported by Shuto group [20, 21]. In this synthesis, treatment of 2’-ketouridine 9 with Lithium bis (trimethylsilyl) amide (LiHMDS) and N-F fluorination agents such as Selectfluor or NFSI gave anomeric mixture of the 1’-fluoro-2’-ketouridine derivatives 10 and (Arabino type congener) in 57-88 % yield. Reduction of the 2’-keto group of 10 by diisobutylaluminiumhydride (DiBAL-H) followed by acetyl protection of the resulted hydroxyl group with acetic anhydride (Ac2 O) and 4-dimethylaminopyridine (DMAP) gave the 1’-fluorouridine derivatives 11 and 12 (8:9=1:4) in 68 % yield. This method has three drawbacks:
1. Only protected 1’-fluorouridine derivatives 11 and 12 were isolated and neither isolation nor deprotection and purification of the free 1’-fluoronucleoside was successful. This is may be due to the instability of the deprotected 1’-fluoruridine.
2. This method gives anomeric mixture of protected 1’-fluoruridines and that leads to low yield of the preferred β-anomer.
3. This method was not applied to purine nucleosides.
2’- Fluorinated nucleosides
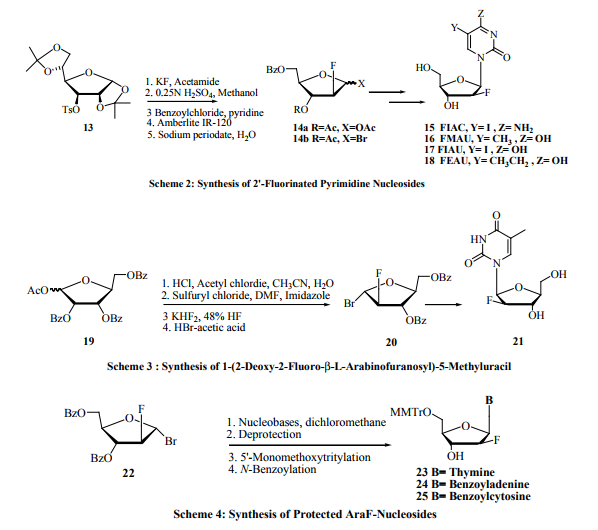
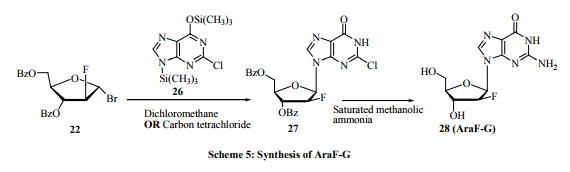
More attention was given to the synthesis of 2’-β-Dfluoronucleosides (araF-nucleosides) over the 2’-α-D-fluoro nucleosides (riboF-nucleosides). This is because of the interesting antiviral activity shown by introducing a fluorine atom at the 2’-β-positions of the nucleosides and nucleoside analogs rather than at 2’-α-positions. Reichmann et al. [23] and Watanabe et al. [24] developed a synthetic method, in two steps, to obtain 2-deoxy-2-fluorD-arabinose 14a and 1-bromo-2-deoxy-2-fluoro-D-arabinose 14b from a readily available D-glucose derivative 13 (Scheme 2). Arabinose sugars 14a-b were used to synthesize series of fluorinated nucleosides 15-18 in a large scale for biological evaluation. Straightforward method for the synthesis of FIAC and FMAU nucleosides was also developed by Tann etal. [25]. For example, 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)- 5-iodocytosine (FIAC, 15), 1-(2-deoxy-2-fluoro-β-Darabinofuranosyl)-5-methyluracil (FMAU, 16), 1-(2-fluoro2-deoxy-β-D-arabinofuranosyl)-5-iodo uracil (FIAU, 17), and 1-(2-fluoro-2-deoxy-β-D-arabinofuranosyl)-5-ethyl uracil (FEAU,18) have shown potent activity against Herpes Simplex Virus (HSV), and excellent activity against hepatitis-B virus (HBV) and cytomegalovirus (CMV). FMAU, 16 have also shown significant activity against murine leukemia [22]. Chu’s group [26, 27] has also reported an efficient procedure for the synthesis of 1- (2-deoxy-2-fluoro-β-Larabinofuranosyl)-5-methyluracil 21 from sugar 19 (Scheme 3). Another interesting approach is the synthesis of the 2’-fluorinated nucleosides, namely 2’-deoxy-2’-fluoro arabinonucleosides (araF-nucleosides) [28, 29]. They are used as building blocks for the synthesis of 2’-deoxy-2’-fluoro arabinonucleic acid (2’-F’ANA), [30-33] a very promising antisense oligonucleotides. Here the 2’-fluoro nucleosides were prepared via condensation of the silylated nucleic acids bases such as silylated N-acetyl cytosine or silylated thymine or N-benzoylated adenine with 2-deoxy-2-fluoro-3, 5-di-Obenzoyl-α-D-arabinofuranosyl bromide 22. Deprotection of the produced nucleosides, followed by 5’-tritylation and N-benzoylation give araF- nucleosides 23-25 in high yield (Scheme 4). The black sheep of this family of 2’-fluoro nucleosides is the 9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl) guanine (araFG) because the above mentioned procedure yields araF-G in low yields. An efficient synthesis of araF-G has been reported by Elzagheid et al. [29]. It involves coupling of 2-deoxy-2-fluoro-3, 5-di-O-benzoyl-α-D-arabinofuranosyl bromide 22 with silylated 2-chloro hypoxanthine 26 to afford 2-chloro-β-araF-I 27 that was transformed in araF-G 28 by treatment with methanolic ammonia in high yield (Scheme 5). Another efficient approach for the synthesis of araF-G 28 was reported by Sivets [34]. The synthesis involves reaction of potassium salts of 2-amino-6-chloropurine with 2-deoxy2-fluoro-3, 5-di-O-benzoyl-α-D-arabinofuranosyl bromide 22. In addition to methanolic ammonia, 2-mercaptoethanol and sodium methoxide in methanol was also used to convert the masked nucleoside to araF-G. In addition to the above mentioned syntheses, a novel synthesis and evaluation of the 5-ethyl analogs of the 2-deoxy-2-fluoro- β -D-arabinofuranosyl nucleosides was reported by Shakya et al. [35]. Among the tested fluorinated nucleosides, the 1-(3-bromo-2, 3-dideoxy-2-fluoro-β-Darabinofuranosyl)-5-ethyluracil has showed promising activity against Mycobacterium tuberculosis and Mycobacterium bovis with no cellular toxicity upto the highest concentration tested (CC50 > 100 µg/mL). A linear synthesis of 2’-deoxy-2’, 2’-difluorocytidine (Gemcitabine) was also reported by Brown et al. [36]. Gemcitabine under the trade name Gemzar, by Lilly Company, was widely used as anticancer drug. This linear synthesis involves conversion of 3, 5-di-O-benzoyl-2-deoxy-2, 2-difluororibose to corresponding glycosyl urea followed by conversion to cytosine base thru the uracil derivative.
3’- Fluorinated nucleosides
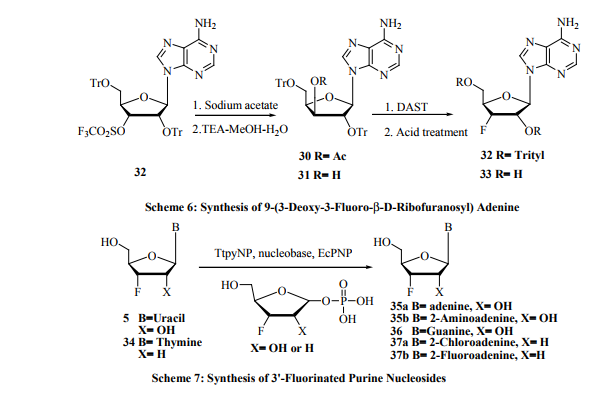
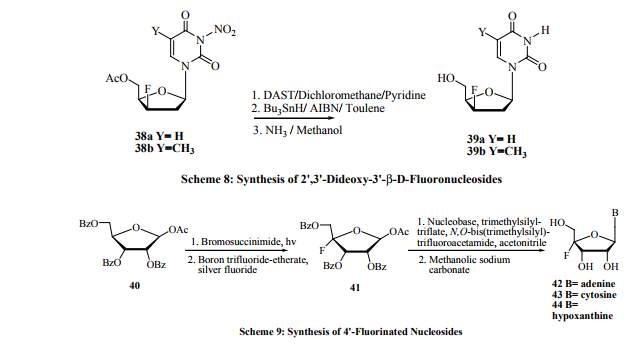
Carbon-3’-fluoronucleosides have shown a wide range of biological activity [37]. Among them, 3’-deoxy-3’-β-Dribofuranosides of adenine, guanine and their 2-deoxy analogs have shown potent antiviral and cytostatic activities [38]. Synthesis of 3’-fluorinatednucleosides such as 9-(3-deoxy3-fluoro-β-D-xylofuranosyl) adenine 1 [15] and 9-(3-deoxy3-fluoro-β-D-arabinofuranosyl) adenine 2 [16](Figure 2) by reacting 1-(5-O-benzoyl-2,3-epoxy-β-D-lyxofuranosyl) adenine with tetraethylammonium fluoride and 1-(5-O-benzoyl2,3-epoxy-β-D-ribofuranosyl) adenine with potassium hydrogen fluoride respectively are subject to few drawbacks. Vigorous reaction conditions are required for the ring opening and strictly anhydrous conditions are needed for the reagent and also to avoid formation of side products. Epoxide ring opening is relatively easy with HF in the presence of tetrahydrofuran or dioxane. This is because of the increased dissociation of the hydrofluoric acid [39]. An alternative route has been reported by Pankiewicz and Watanabe [40] for the synthesis of 9-(3-deoxy-3-fluoro-β- D-ribofuranosyl) adenine 33. This involves the treatment of the nucleoside triflate 29 with sodium acetate to give 3’-Oacetyl derivative 30. Mild hydrolysis of the later nucleoside in triethylamine-methanol-water mixture gave nucleoside 31 in good yield. Further treatment with DAST [(diethylaminosulfurtrifluoride, Et2 NSF3 ] gave the desired 3’-fluorosubstituted nucleoside 32 in high yield. Acidic treatment of 32 with acid gave nucleoside 33 (Scheme 6). Another interesting approach is the synthesis of 3’-fluorinated purine nucleosides 35-37 (Scheme 7) from 3’-fluorinated pyrimidine nucleosides 5 and 34 with the application of recombinant T. thermophilus pyrimidine nucleoside phosphorylase (TtpyNP) and E-Coli purine nucleoside phosphorylase (EcPNP) [41]. Another straightforward synthesis of 2’, 3’-dideoxy-3’- β-fluoronucleosides was reported by Khalil et al.42 The 5’-acetyl-3-nitro-2’-deoxynucleodies 38a-b were reacted with DAST in anhydrous dichloromethane-pyridine mixture to give the corresponding 3’- β -fluoronucleosides. Deprotection of the 3-nitro group was achieved by tributyltin hydride (Bu3SnH) in dry toluene in the presence of α, α’- azoisobutyronitrile (AIBN). Removal of the 5’-acetyl with methanolic ammonia afforded nucleosides 39a-b (Scheme 8).
4’- Fluorinated nucleosides
Another class of the fluoronucleosides that has attention of late is the 4’-fluoronucleosides. They are expected to have significant values in a variety of biochemical studies. A fruitful procedure has been reported by Lee et al. [19] where various 4’-fluorinated nucleosides 42-44 have been prepared in three steps via sequential bromination and fluorination of the ribofuranose 40. Glycosylation of the 4-fluoro-β-Dribofuranose 41 with N, O-bis-(trimethylsilyl) trifluoroacetamide and trimethylsilyl triflate followed by debenzoylation gave the desired fluoronucleosides in good yield (Scheme 9).
5’- Fluorinated nucleosides
Four approaches for the synthesis of 5’-fluoronucleosides 45-48 (Figure 3) have been reported. These include:
1. Nucleophilic displacement of sulfonates by fluoride ions: This reaction is carried out in either mesylates or tosylates with potassium fluoride in ethylene glycol or with tetrabutylammonium fluoride in dimethylformamide (DMF). However, the use of hydrofluoric acid in dioxane has been reported to give better yields [17].
2. Opening of the O2 , 5’-bond of anhydronucleosides: these anhydronucleosides can serve as good starting materials but they are rarely used [43].
3. Reaction of a free 5’-hydroxyl group with DAST. This reagent has been successfully applied for the replacement of hydroxyl group by a fluorine atom [44].
4. Displacement of iodine as leaving group: In this approach silver fluoride in pyridine has been used for introduction of fluorine atom at 5’-position of the 5’-iodouridine [14].
CONCLUSIONS
In this review, a highlighted number of important synthetic methods of biologically active fluorinated nucleosides have been covered. A fluorine atom, as a mimic of hydrogen or hydroxyl group, that has been introduced in different positions in the sugar moiety of the nucleoside has surely improved their pharmacological and biological properties.
ACKNOWLEDGMENT
Author acknowledges the immense help received from the scholars whose articles are cited and included in references of this manuscript. The author is also grateful to authors / editors / publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed.
References:
1. Turner M A, Yang X, Yin D, Kuczera K, Borchardt R T, Howell P L. Structure and function of S-adenosylhomocysteine hydrolase. Cell Biochem Biophys 2000; 33: 101-125.
2. Kitade Y, Kozaki C, Miwa T, Nakanishi M. Synthesis of basemodified noraristeromycin derivatives and their inhibitory activity against human and plasmodium falciparum recombinant S-adenosylhomocysteine hydrolase. Tetrahedron 2002; 58: 1271-1277.
3. Anderson K S. Perspectives on the molecular mechanism of inhibition and toxicity of nucleoside analogs that target HIV-1 reverse transcriptase. Biochimica et Biophysica Acta 2002; 1587: 296-299.
4. Welch J T. Advances in the preparation of biologically active organofluorine compounds. Tetrahedron 1987; 43 (14): 3123- 3197.
5. Wilkinson J A. Recent advances in the selective formation of the carbon-fluorine bond Chem Rev 1992; 92 (4): 505-519.
6. Purrington S T, Kagen B S, Patrick T B. Application of elemental fluorine in organic synthesis. Chem Rev 1986; 86 (6): 997- 1018.
7. Schlosser M. Introduction of fluorine into organic molecules: why and how. Tetrahedron 1978; 34 (1): 3-17.
8. Ma T, Lin J-S, Gary N M, Cheng Y-C, Chu C K. Synthesis and anti-hepatitis B virus activity of 9-(2-deoxy-2-fluoro-β-larabinofuranosyl) purine nucleosides. J Med Chem 1997; 40: 2750–2754.
9. Kalota A, Karabon L, Swider CR, Viazovkina E, Elzagheid MI, Damha MJ, and Gewirtz AM. 2’-Deoxy-2’-fluoro-β-Darabinonucleic acid (2’F-ANA) modified oligonucleotides (ON) effect highly efficient, and persistent, gene silencing. Nucleic Acids Research 2006; 34 (2): 451–461.
10. Takamatsu S, Katayama S, Hirose N, De Cock E, Schelkens G, Demillequand M, Brepoels J, and Izawa K. CONVENIENT SYNTHESIS OF FLUORINATED NUCLEOSIDES WITH PERFLUOROALKANESULFONYL FLUORIDES. Nucleosides Nucleotides and Nucleic Acids 2002; 21(11-12):849-861.
11. Yamada K, Matsumoto N, and Hayakawa H. Stereoselective Synthesis of 2-Deoxy-2-Fluoroarabinofuranosylα-1-Phosphate and Its Application to the Synthesis of 2′-Deoxy-2′-Fluoroarabinofuranosyl Purine Nucleosides by a Chemo-Enzymatic Method. Nucleosides Nucleotides and Nucleic Acids 2009; 28 (11-12): 1117-1130.
12. Pankiewicz K W. Fluorinated nucleosides. Carbohydr Res 2000; 327: 87-105.
13. Kodama T, Matsuda A, Shuto S. The first synthesis of 1’-fluoronucleosdies. Nucleic Acids Symposium Series 2006; 50: 3-4.
14. Herdewijn P, Van Aerschot A, Kerremans L. Synthesis of nucleosides fluorinated in the sugar moiety. The application of diethylaminosulfur trifluoride to the synthesis of fluorinated nucleosides. Nucleosides, Nucleotides 1989; 8(1): 65-96.
15. Robins M J, Fouron Y, Mengel R. Adenosine 2′, 3′-ribo-epoxide. Synthesis, intramolecular degradation, and transformation into 3′-substituted xylofuranosyl nucleosides and the lyxo-epoxide. J Org Chem 1974; 39: 1564-1570.
16. Miyai K, Robins RK, Tolman RL. Synthesis of 9-(3-deoxy-3- fluoro-β-D-arabinofuranosyl) adenine. J Med Chem 1972; 15: 1092-1994.
17. Kissman HM, Weiss MJ. 5-Deoxy-5-fluoro-D-ribofuranosyl Derivatives of Certain Purines, Pyrimidines and 5, 6-Dimethylbenzimidazole. J Am Chem Soc 1958; 80 (20): 5559–5564.
18. Kowollik G, Gaertuer K, Langen P. Nucleosides of fluorocarbohydrates. XIII. Synthesis of 3’ deoxy 3’ fluorouridine. J. Carbohydr Nucl Nucl 1975; 2 (3): 191-195.
19. Lee S, Uttamapinant C, Verdine G L. A concise synthesis of 4’-fluoronucleosides. Org Lett 2007; 9 (24): 5007-5009. And references therein.
20. Kodama T, Matsuda A, Shuto S. Synthesis of 1’-fluorouracil nucleosides as potential antimetabolites. Tetrahedron 2006; 62: 10011-10017.
21. Kodama T, Shuto S, Matsuda A. Synthesis of pyrimidine 1’-fluoronucleosides. Tetrahedron Lett 2006; 47: 4429-4432.
22. Liu P, Sharon A, Chu CK. Fluorinated nucleosides. J Fluor Chem 2008; 129 (9): 743-766.
23. Reichmann U, Watanabe KA, Fox JJ. A practical synthesis of 2-deoxy-2-fluoro-D- arabinofuranose derivatives. J Carbohydr Res 1975; 42 (2): 233-240.
24. Watanabe KA, Su TL, Klein RS, Chu CK, Matsuda A, Chun MW, Lopez C, Fox JJ. Synthesis of antiviral nucleosides: 5-substituted 1-(2-deoxy-2-halogeno-β-D-arabinofuranosyl) cytosine and -uracil. Some structure-activity relationships. J Med Chem 1983; 26 (2): 152-156.
25. C. H. Tann, P. R. Brodfuehrer, S. P. Brundidge, C. Sapino, H. G. Howell, Fluorocarbohydrates in synthesis. An efficient synthesis of 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-5-iodouracil (β-FIAU) and 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl) thymine (β -FMAU). J Org Chem 1985; 50: 3644-3647.
26. Ma T, Pai SB, Zhu XL, Lin JS, Shanmu K, Du J, Wan C, Kim H, Newton MG, Cheng YC, Chu CK. Structure--activity relationships of 1-(2-Deoxy-2-fluoro-β-L arabinofuranosyl)pyrimidine nucleosides as anti-hepatitis B virus agents. J Med Chem 1996; 39 (14): 2835-2843.
27. Du J, Choi Y, Lee K, Chun BK, Hong JH, Chu CK, A Practical Synthesis of L-FMAU from L-Arabinose. Nucleosides Nucleotides 1999; 18: 187-192.
28. Elzagheid M I, Viazovkina E, Damha M J. Synthesis of protected 2’-deoxy-2’-fluoro-β-D-arabinonucleosides.Current Protocols in Nucleic Acid Chemistry 2002; 1.7.1–1.7.19. And references therein.
29. Elzagheid M I, Viazovkina E, Damha M J. A new synthesis of 9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl) guanine. Nucleosides, Nucleotides and Nucleic Acids 2003; 22: 1339-1342. And references therein.
30. Damha M J, Wilds C J, Noronha A, Brunker I, Brokow G, Arion D, Parniak M A. Hybrids of RNA and arabinonucleic acids (ANA and 2’F-ANA) are substrates of ribonuclease H. J AmChem Soc 1998; 120 (49): 12976-12977.
31. Wilds C J, Damha M J. 2’-Deoxy-2’-fluoroarabinonucleosides and oligonucleotides (2’F-ANA): synthesis and physicochemical studies. Nucleic Acids Res 2000; 28: 3625–3635.
32. Lok C N, Viazovkina E, Min K L, Nagy E, Wilds C J, Damha M J, Parniak M A. Potent gene-specific inhibitory properties of mixed-backbone antisense oligonucleotides comprised of 2’-deoxy-2’-fluoro-D-arabinose and 2’-deoxyribose nucleotides. Biochemistry 2002; 41 (10): 3457-3467.
33. Viazovkina E, Mangos M M, Elzagheid M I, Damha M J. solid-phase synthesis of 2′-Deoxy-2′-fluoro-β-Doligoarabinonucleotides (2′F-ANA) and their phosphorothioate derivatives. Current Protocols in Nucleic Acid Chemistry 2002; 4.15.1-4.15.22.
34. Sivets G G. Syntheses of 2’-deoxy-2’-fluoro-β-Darabinofuranosyl purine nucleosides via selective glycosylation reactions of potassium salts of purine derivatives with glycosyl bromide. Tetrahedron Lett 2016; 57: 268-271.
35. Shakya N, Srivastav N C, Bhavanam S, Tse C, Desroches N, Agrawal B, Kunimoto D Y, and R. Kumar R. Discovery of novel 5-(ethyl or hydroxymethyl) analogs of 2’-‘up’ fluoro (or hydroxyl) pyrimidine nucleosides as a new class of Mycobacterium tuberculosis Mycobacterium bovis and Mycobacterium Avium inhibitors. Bioorganic and Med Chem 2012; 20: 4088-4097.
36. Brown K, Weymouth-Wilson A, and Linclau B. A linear synthesis of gemcitabine. Carbohydrate Res 2015; 406: 71-75.
37. Qui X-I, Xu X-H, and Qing F-L. Recent advances in the synthesis of fluorinated nucleosides. Tetrahedron 2010; 66: 789-843.
38. Mikhailopulo I A, Poopeiko N E, Prikota T I, Sivets G G, Kvasyuk E I, Balzarini J, De Clercq E. Synthesis and antiviral and cytostatic properties of 3′-deoxy-3′-fluoro- and 2′-azido-3′-fluoro2′,3′-dideoxy-D-ribofuranosides of natural heterocyclic bases. J Med Chem 1991; 34 (7): 2195-2202.
39. Hirschman R E, Miller R, Wood J, Jones R E. The Reaction of Epoxides with Anhydrous Hydrogen Fluoride in the Presence of Organic Bases. The Preparation of 9-α-Fluoro-4-pregnene-11β, 17α, 21-Triol 3, 20-Dione 21-Acetate and Its 1-Dehydro Analog. J Am Chem Soc 1956; 78 (19): 4956-4959.
40. Pankiewicz K W, and Watanabe K A. A synthesis of 2’-fluoroand 3’-fluoro-substituted purine nucleosides via a direct approach. Nucleosides and Nucleotides as Antitumor and Antiviral Agents, Plenum Press, New York, 1993; 55-71.
41. D. V. Burko, Biotechnology in Medicine, Food Stuffs, Biocatalysis, Environment and Biogeotechnology, eds S. D. Varfolomeev, G. E. Zaikov, L. P. Krylova. Nova Science Publisher, New York, 2010; 1-13.
42. Khalil A, Mathe C, and Perigaud C. Rapid synthesis of 2’,3’-dideoxy-3’-β-fluoro-pyrimidine nucleosides. Bioorganic Chem 2010; 38: 271-274.
43. Kowollik G, Schutt M, Langen P, Demirow G. Nucleoside von Fluorzuckern; Synthese der 5′-Desoxy-5′-fluor-Derivate von 6-Aza-uridin und Ribofuranosyl-thymin. Z Chem 1972; 12 (3): 106-107.
44. Middleton W J. New fluorinating reagents. Dialkylaminosulfur fluorides. J Org Chem, 1975; 40 (5): 574-578.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





