IJCRR - 10(3), February, 2018
Pages: 19-27
Date of Publication: 01-Feb-2018
Print Article
Download XML Download PDF
Virtual Screening and Docking Analysis of Novel Flavonoidanalogues as Antipsoriaticagents
Author: Parakh Sharma, Rohit Mishra, Babu Vakil
Category: Healthcare
Abstract:Introduction: Psoriasis is an immune-mediated chronic, inflammatory skin disease characterized by hyper proliferative keratinocytes and their infiltration into the dermis of T cells, dendritic cells, macrophages and neutrophils. There is ample evidence suggesting the key role of dysregulation of immune cells in the skin, particularly T cells, in the pathogenesis of psoriasis. Calcineurin, a calcium and calmodulin dependent serine/threonine protein phosphatase plays a major role in increasing the production of T cells and thus in the development of psoriasis. Flavonoids such as quercetin and kaempferol are known to inhibit development of psoriasis.
Objective: We have performed docking of novel designed flavonoid analogues of quercetin and kaempferol to Calcineurin by insilicoanalysis to predict their potential as antipsoriatic candidates.
Materials: Eighty analogues each of quercetin and kaempferol were designed using Schrödinger- Maestro 11 and docked with Calcineurin using PyRx software.
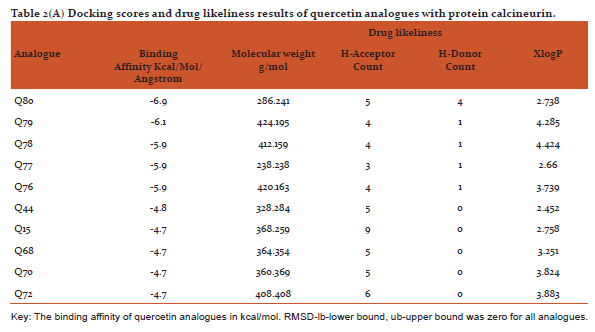
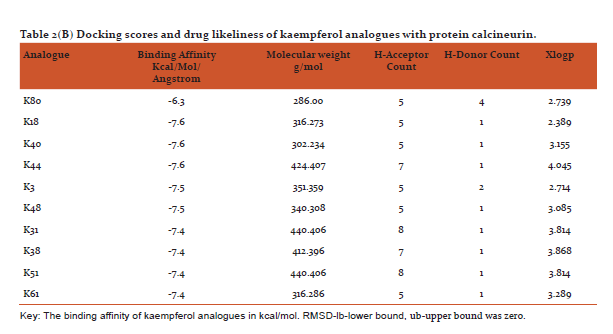
Results: The best binding affinities (kcal/mol) were predicted for5 quercetin analogues- Q79 (-6.1), Q78, Q77 andQ76 (-5.9) and Q44 (-4.8) and5 kaempferol analogues-K18, K40 andK44 (-7.6), K3, K48 (-7.5).
Conclusion: This study using PyRx software strongly supports the importance of computational approach in drug discovery.
The short listed novel analogues of quercetin and kaempferol follow Lipinski rule of 5, satisfying basic parameters for drug likeliness. QSAR and pharmacokinetic analysis can be deployed in future, to further characterize them. Eventually, most promising analogues can be synthesized and evaluated to verify their actual antipsoriatic activity.
Keywords: Psoriasis, Calcineurin, Kaempferol, Docking, PyRx software
DOI: 10.7324/IJCRR.2017.1035
Full Text:
INTRODUCTION
Psoriasis is an immune-mediated chronic, inflammatory skin disease characterized by hyperproliferative keratinocytes and infiltration of T cells in the dermis.[1]Advances in understanding the immune-mediated pathological mechanisms of psoriasis have opened new therapeutic avenues. [2] There are many possible protein targets in psoriasis which can be inhibited and Calcineurin (CaN) is a popular target to suppress the activation of memory CD4+T cells and their proliferation that plays an important role in psoriasis.[3]A class of drugs called calcineurin inhibitors which include compounds like cyclosporin, voclosporin and tacrolimus are already in use clinically.[4] Cyclosporin A (CsA) is known to bind to Nuclear factor of activated T cells (NF-ATc).[5,6] Thus, blocked NF-ATc transport in nucleus, in turn, blocks T-cell pathway leading to antipsoriatic activity.[7] However, extensive use of CsA is restricted by its severe side effects.[8,9]
Herbal drugs are known to have lesser side effects, ease of availability and may lend themselves as potential antipsoriatic moieties.[10] Flavonoids, have been the subject of extensive research and a variety of compounds showing beneficial effects, have been identified.[11,12] Quercetin, a flavonol, has been reported to inhibit the activity of Calcineurin in Human Embryonic Kidney cells 293 (HEK293).[13] Kaempferol, another flavonol, has also been identified as a novel calcineurin inhibitorin Jurkat cell line model.[14] Therefore, there is for exploring such flavonoids and their analogues for more potent antipsoriatic activity.
Virtual screening(VS), a process of computationally analyzing large compound libraries, to discover new drugs, has become increasingly popular tool in drug discovery research wherein the number of methods and software which use the ligand/target approach is increasing at a rapid pace.[15] The energy function that evaluates the binding free energy between protein and ligand is known as a scoring function. The steps include protein structure preparation, ligands database preparation and docking calculations.[15,16] Determining protein surface atoms and site points as well as assignment of interaction data are sometimes, internally included in the docking software like in PyRx, Schrödinger-Maestro 11 or Discovery Studio.[17]The best ligand hits are predicted by computational approaches that ‘dock’ library of ligands into the structures of target proteins and ‘score’ their potential complementarities to binding sites.[18]
The objective of the present work was to predict promising new flavonoid analogues of kaempferol and quercetin as antipsoriatic agents that will specifically bind to the target protein
calcineurin.[19]The study was performed using Maestro-11 for docking analysis.[15,17,20] The most promising predicted analogues then can be synthesized and evaluated using in vitro/in vivo testing methods. Currently there are very few effective drugs available for treatment of debilitating and life devastating disease psoriasis and work reported here is an attempt to fulfill the urgent need to identify more potent analogues of phytochemicals.[20]
MATERIALS AND METHODS
Selection of Calcineurin protein and optimization Protein calcineurin Protein Data Bank (PDB)ID: 1MF8 was imported from the PDB (http://www.rcsb.org/pdb) [21] andwas analyzed for its active site by discovery studio visualizer (http://accelrys.com).[22,23,24] The protein preparation wizard was used to correct the imported protein coordinates using Maestro-GUI (Schrödinger Suite).[25,26]The corrections involved adding hydrogen atoms, defining the bond orders, deleting unwanted water molecules and salts followed by optimizing hydrogen bond network. All the atoms in the system were atom-typed using the OPLS2005 force field.[26,27]
The protonation states of titratable amino acid residues were defined at pH 7.4 (physiological state) using PropKatoolin- Schrödinger Suite. For minimizing the protein in explicit water box, appropriate numbers of TIP3P water molecules along with number of ions were added to neutralize the system using Desmond module.
The protein coordinates were optimized in 3 minimization cycles, consisting of 100,000 steps with convergence criteria of 0.001 kcal/mol/Angstrom. First 1000 steps employed steepest descents minimizer and remaining steps used Conjugate- gradients minimizer.[28] The first cycle of minimization placed 25 kcal/mol/Angstrom^2 force constant. This force was reduced to 5 kcal/mol/angstrom^2 and the last cycle of minimization was performed without any restraining potentials for final energy minimization. If the system is unable to relax without any restrains after the last cycle then it is an indication that the system is not a good starting point.[29,30]
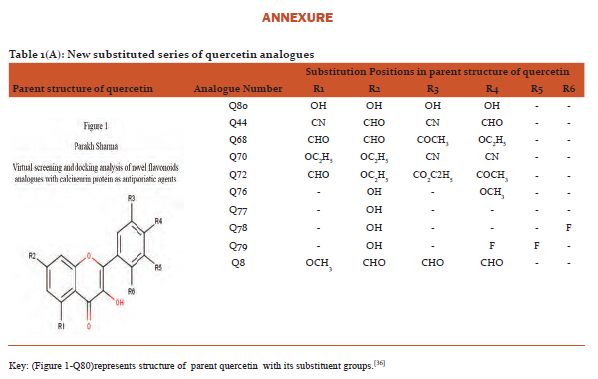
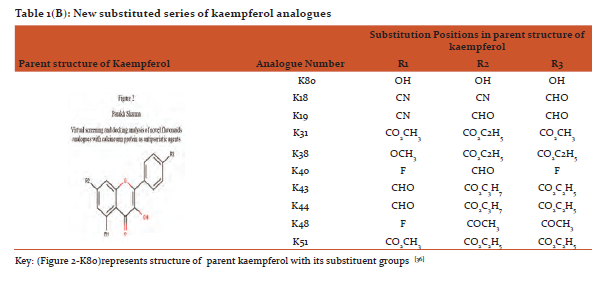
Designing and preparation of analogues: The basic structures of quercetin and kaempferol are shown in figure 1and2. The structures were retrieved from Pub Chem database (https://pubchem.ncbi.nlm.nih.gov/).[31]Theirnovel analogues were designed by maestro-11 and saved in MDL files (V2000)and were converted to SDF files used as input in PyRx software.(http://PyRx.sourceforge.net/).[32]
Docking analysis and grid generation: Open Babel tool was used for optimizing designed analogues with forcefields MMFF94 and GHEMICAL. Analogues energy was minimized by Conjugate Gradient and Steepest Descent optimization algorithms. The cycle consisted of 50000 steps with convergence criteria of 0.001 kcal/mol/Angstrom. Finally ‘all ligand’ option was used to convert the minimized files to PDBQT format to generate their atomic coordinates for docking. For docking analysis, the protein1MF8 grid box was set at the retained coordinates of cyclosporin A where center_x = -37.0035159475, center_y = 16.4330071521, and center_z = 24.2216177997.[33] Drug likeliness parameters were predicted by VLIFEMDS-QSARPro (www.vlifesciences. com).[34,35] The best interactions were visualized by Discovery Studio.[22]
RESULTS
The data for parent structure of quercetin (Figure 1-Q80) and its novel designed analogues with functional groups substitution is presented in table 1(A) whereas parent structure of kaempferol (Figure 2-K80)and data for its novel designed analogues are shown in table1(B).[36]
Table 2(AandB) shows docking scores (kcal/mol/Angstrom) retrieved from PyRx software along with parameters of drug likeliness (hydrogen bond donor/acceptor, xlogp and molecular weight) using VLIFEMDS-QSAR Pro. The PyRx software uses a measure of distance between the experimental and predicted structures to compare the accuracy of the predictions. Root mean square deviation are calculated relative to the best mode and using only movable heavy atoms. Two variants of RMSD are lb-lower bound and ub-upper bound, differing in how the atoms are matched in the distance calculation.[32]
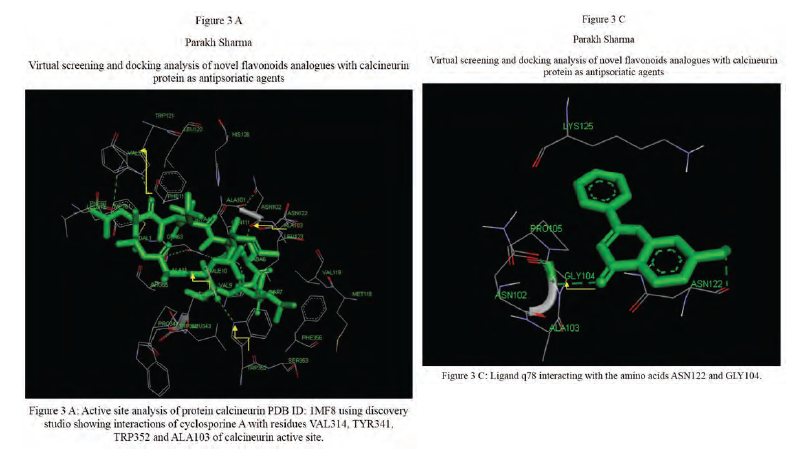
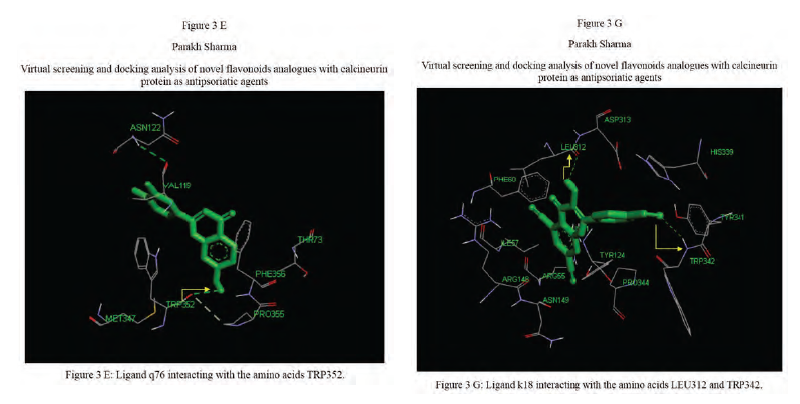
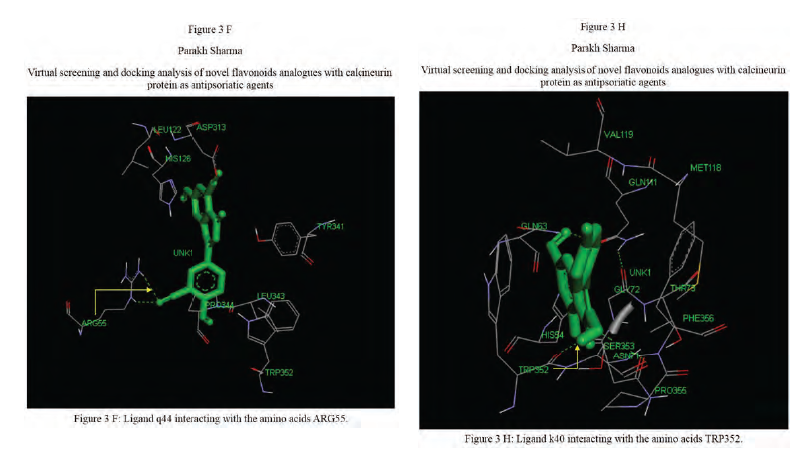
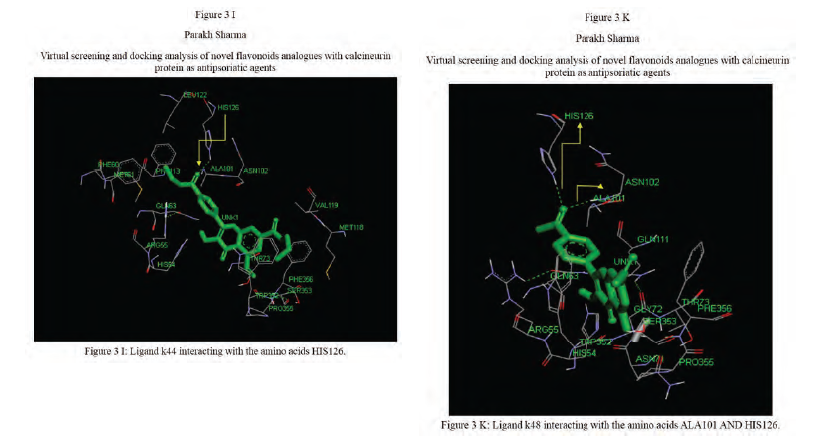
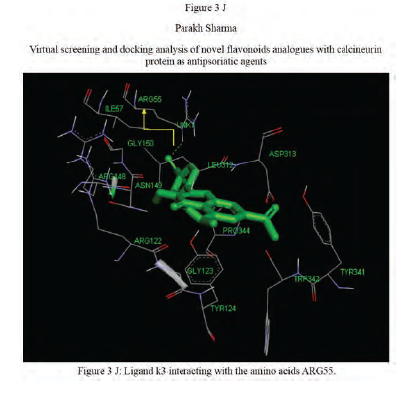
Calcineurin protein PDB ID: 1MF8 active site analysis by discovery studio software is depicted in figure3(A)where amino acid residues val314, ala103,tyr341 and trp 352 are interacting with the known antipsoriatic drug cyclosporin A. Next, the newly designed analogues of quercetin and kaempferol from table 1 AandB were then docked at the same active site of calcineurin to predict best analogues binding mode.[33] Top scored quercetin analogues interactions with protein ID: 1MF8 are shown in Figure 3 (B-F) similarly Figure3 (G-K) shows interactions of best kaempferol analogues with 1MF8.
DISCUSSION
Some in vitro studies have shown that flavonoids like quercetin and kaempferol have anti-psoriatic activity against the calcineurin protein.[37] Such reports have shown that best docked analogues of quercetin and kaempferol do have favorable ligand-protein molecular interactions, similar to the interaction of cyclosporin A with calcineurin.[33] In our present docking analysis, datasets of 80 analogues each of kaempferol and quercetin flavonoids were docked at the active site of calcineurin protein using PyRx software. The docking data of ligand-protein molecular interactions for 5 best quercetin analogues namely Q79 (-6.1), Q78, Q77 and Q76 (-5.9) and Q44 (-4.8) and for best 5 kaempferol analogues namely K18,K40 andK44 (-7.6) and K3andK48 (-7.5)are depicted in figure 3(B-F) and (G-K), respectively.
Kartasasmita RE et al, (2010) have performed docking study using Arguslab software on inducible nitric oxide synthase (PDB ID: 1M9T) with quercetin derivatives. They predicted
more potent iNOS inhibitors than parent quercetin, asQuercetin- 3-O-acetate with -10 kcal/mol and 6,8-dichloroquercetin- 3-O-acetate with -7.49 kcal/mol binding score.[38]
Zaveri et al (2015) showed that the target protein- Deltalactam- biosynthetic de-N-acetylase protein (PDB ID: 2J13), when docked with potential ligands taken from NCI and Drug Bank databases, the best scored ligands were identified as NCI-293778 and Rofecoxib which share the common site residues as that of residues predicted in CASTp tool binding pocket predictor. [33]
From the drug likeliness analysis shown in table 2 (AandB) it becomes evident that the best analogues are in the acceptable range by Lipinski rule of 5[35] and may have similar pharmacological properties like parent structures of quercetin and kaempferol. It is hoped that some of them may possess better antipsoriatic activity.[3,5] Dash R (2015) has revealed that when docking was performed with COX-2 protein and the 12 natural flavonoids compounds, afavorable binding energy of > -8 kcal/mol in ArgusLab docking software was predicted [39]Sharma and Vakil(2017)using2D QSAR method by VLIFEMDS-QSARpro software, have predicted antipsoriatic activity for some novel analogues of quercetin and kaempferol.[40]
In line with the above discussion related to usefulness of docking studies, our work, using PyRx software, has predicted and confirmed that some novel analogues of quercetin which show similar binding scores as parent structure and kaempferol analogues too have better binding score compared to parent structure. These analogues do show favorable
drug likeliness properties and bind to the same active site of target protein calcineurin where drugs like cyclosporin A are known to bind.
CONCLUSION
This study performed with PyRx software strongly supports the importance of computational approach in early part of drug discovery research. It results in saving enormous amount of time, resources and money. The novel quercetin and kaempferol analogues follow Lipinski rule of 5. In future, 3D QSAR studies coupled with pharmacokinetic analysis can validate predicted potential of these antipsoriatic agents. Most promising analogues can be synthesized in vitro and tested on human keratinocyte cell lines and if results are satisfactory, it may prove to be rewarding experience in terms of finding more potent antipsoriatic agents.
ACKNOWLEDGMENT
Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to authors / editors / publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed.
Source of Funding: External funding –Nil. Internally .financed by the college

References:
1. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cellular and molecular immunology. 2012 Jul 1;9(4):302-9.
2. Krueger JG, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Annals of the rheumatic diseases. 2005 Mar 1;64(2):30-36.
3. Sieber M, Baumgrass R. Novel inhibitors of the calcineurin/ NFATc hub-alternatives to CsA and FK506?. Cell Communication and Signaling. 2009 Oct 27;7(1):25-44.
4. Khurana A, Brennan DC. Current concepts of immunosuppression and side effects. In Pathology of solid organ transplantation 2009. Springer Berlin Heidelberg.2(1):11-30.
5. Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes and development. 2003 Sep 15;17(18):2205-2232.
6. Bronstein-Sitton N. T cell signalling and activation: no simple matter. Pathways. 2006;2:8-11. 7. Tedesco D, Haragsim L. Cyclosporin: a review. Journal of transplantation. 2012 Jan 4;(2012):1-7.
8. Berger TG, Duvic M, Van Voorhees AS, Frieden IJ. The use of topical calcineurin inhibitors in dermatology: safety concerns. Journal of the American Academy of Dermatology. 2006 May
1;54(5):818-823.
9. Johnson-Huang LM, Lowes MA, Krueger JG. Putting together the psoriasis puzzle: an update on developing targeted therapies. Disease models and mechanisms. 2012 Jul 1;5(4):423 433.
10. García-Pérez ME, Stevanovic T and Poubelle PE . New therapies under development for psoriasis treatment. Current opinion in pediatrics,2013; 25(4):480-487.
11. Geetha M. Anti-psoriatic activity of flavonoids from Cassia tora leaves using the rat ultraviolet B ray photodermatitis model. RevistaBrasileira de Farmacognosia. 2014 Jun;24(3):322-329.
12. Vijayalakshmi A, Geethab M and Ravichandiran V. Anti-Psoriatic Activity of Flavonoids from the Bark of Givotiarottleriformis Griff. Ex Wight. Iranian Journal of Pharmaceutical Sciences, 2014; 10 (3): 81- 94.
13. Zhao X, Wang Q, Yang S, Chen C, Li X, Liu J, Zou Z, Cai D. Quercetin inhibits angiogenesis by targeting calcineurin in the xenograft model of human breast cancer. European journal of pharmacology. 2016 Jun 15;781:60-68.
14. Kaidama WM, Gacche RN. Antiinflammatory activity of Quercetine in acute and chronic phases of inflammation in Guinea pigs. American Journal of Phytomedicine and Clinical Therapeutics. 2015 Feb 27;3(2):129-136.
15. Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Current medicinal chemistry. 2013 Aug 1;20(23):2839-2860.
16. JE MA, MN A. Virtual screening and lead optimisation to identify novel inhibitors for HDAC-8. arXiv preprint arXiv: 1209.2793. 2012 Sep 13:1-58.
17. Lionta E, Spyrou G, K Vassilatis D, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Current topics in medicinal chemistry. 2014 Aug 1;14(16):1923-1938.
18. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature reviews Drug discovery. 2004 Nov 1;3(11):935- 949.
19. Varadwaj PK, Sharma A, Kumar R. An overview of psoriasis with respect to its protein targets. Egyptian Dermatology Online Journal. 2010 Jun 1;6(1):1-13.
20. Asgaonkar KD, Mote GD, Chitre TS. QSAR and molecular docking studies of oxadiazole ligated pyrrole derivatives as enoyl-ACP (CoA) reductase inhibitors. Scientia pharmaceutica. 2013 Nov 24;82(1):71-86.
21. Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallographica Section D: Biological Crystallography, 54(6):1078-
1084.
22. Sussman JL, Lin D, Jiang J, Manning NO, Prilusky J, Ritter O, Abola EE. Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallographica Section D: Biological Crystallography. 1998 Nov 1;54(6):1078-84.
23. Jamal QM, Lohani M, Siddiqui MH, Haneef M, Gupta SK, Wadhwa G. Molecular interaction analysis of cigarette smoke carcinogens NNK and NNAL with enzymes involved in DNA repair pathways: An in silico approach. Bioinformation. 2012;8(17):795-800.
24. Kishore DP, Maillabaram R, Rao AR, Rao PM. Antiinflammatory Evaluation and Docking Studies of Some New Thienopyrimidines. Asian Journal of Chemistry. 2013 Dec 11;25(18):10583- 10587.
25. Maestro 11: A drug designing suite of Schrödinger: Available at https://www.Schrodinger.com/maestro 2005. Accessed on 17 August 2017, 2:09 pm.
26. Banerjee K, Gupta U, Gupta S, Wadhwa G, Gabrani R, Sharma SK, Jain CK. Molecular docking of glucosamine-6-phosphate synthase in Rhizopusoryzae. Bioinformation. 2011;7(6):285- 290.
27. Srinivasan P, Chella Perumal P, Sudha A. Discovery of novel inhibitors for Nek6 protein through homology model assisted structure based virtual screening and molecular docking approaches. The Scientific World Journal. 2014 Jan 22;2014:http:// dx.doi.org/10.1155/2014/967873
28. Suvannang N, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Molecular docking of aromatase inhibitors. Molecules. 2011 Apr 28;16(5):3597-617.
29. Jyoti K, Bhatia RK, Martis EA, Coutinho EC, Jain UK, Chandra R, Madan J. Soluble curcumin amalgamated chitosan microspheres augmented drug delivery and cytotoxicity in colon cancer cells: In vitro and in vivo study. Colloids and Surfaces B: Biointerfaces. 2016 Dec 1;148:674-683.
30. S Chintakrindi A, AF Martis E, J Gohil D, T Kothari S, S Chowdhary A, C Coutinho E, A Kanyalkar M. A Computational Model for Docking of Noncompetitive Neuraminidase Inhibitors and Probing their Binding Interactions with Neuraminidase of Influenza Virus H5N1. Current computer-aided drug design. 2016 Dec 1;12(4):272-281.
31. PubChem database, 2004: Available at https://pubchem.ncbi. nlm.nih.gov/, Accessed on 5 August 2017, 11:56 am.
32. PyRx tool: Docking analysis tool,2009: Available at http://PyRx. sourceforge.net/, Accessed on 15 September 2017, 10:52 am.
33. Zaveri K, Chaitanya AK, Reddy IB. Virtual screening and docking studies of identified potential drug target: Polysaccharide deacetylase in Bacillus anthracis. International Letters of Natural Sciences. 2015;7(34):70-77.
34. VLIFEMDS Integrated platform for computer aided drug design (CADD), 2008. Available at : http://www.vlifesciences.com/ products/VLIFEMDS/Product_VLIFEMDS.phph, Accessed on 6 September 2017, 9:15 am.
35. Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technologies. 2004 Dec 31;1(4):337-341.
36. Materska M. Quercetin and its derivatives: chemical structure and bioactivity-a review. Polish journal of food and nutrition sciences. 2008;58(4):407-413.
37. Vijayalakshmi A, Ravichandran V, Velraj M, Nirmala S, Jayakumari S. Screening of flavonoid “quercetin” from the rhizome of Smilax china Linn. for anti–psoriatic activity. Asian Pacific journal of tropical biomedicine. 2012 Apr 1;2(4):269-275.
38. Kartasasmita RE, Herowati R, Gusdinar T. Prediction of their Absorption and Distribution Properties. Journal of Applied Sciences. 2010;10(23):3098-4104.
39. Dash R, Uddin MM, Hosen SZ, Rahim ZB, Dinar AM, Kabir MS, Sultan RA, Islam A, Hossain MK. Molecular docking analysis of known flavonoids as duel COX-2 inhibitors in the context of cancer. Bioinformation. 2015;11(12):543-549.
40. Sharma P K and Vakil B V. Predictive QSAR analysis of flavonoid analogues as antipsoriatic agents. International journal of pharmaceutical sciences and research. 2017; 8(12): 1000-1014.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





