IJCRR - 5(1), January, 2013
Pages: 120-126
Print Article
Download XML Download PDF
CUTANEOUS ANGIOSARCOMA OF FACE - A CASE REPORT
Author: Gunvanti B. Rathod, Atul Jain, Pragnesh Parmar
Category: Healthcare
Abstract:Cutaneous angiosarcoma of the face and scalp is a rare but aggressive vascular tumour of endothelial cell origin, having poor prognosis. Clinically it presents with an asymptomatic erythematous to bruise like macular lesion in face or scalp. Local spread, recurrence and metastasis to regional lymph nodes and lungs are common. Early diagnosis and treatment are essential for local control of this aggressive tumour, but recognition can be delayed because of its rarity or because of difficulty in making a pathological diagnosis. Early detection and multidisciplinary approach may improve survival. Cutaneous angiosarcoma (CA) accounts for 60% of cases of angiosarcoma. We report here a case of 65 years old man who presented with 4 months history of indurated swelling over the left infraorbital area extending the forehead, scalp and the other side of face with marked induration of the skin. Histsopathological examination of the biopsy from the nodule confirmed the diagnosis of cutaneous angisarcoma.
Keywords: Cutaneous angiosarcoma, Vascular tumour, Poor prognosis.
Full Text:
INTRODUCTION
Angiosarcoma is a rare, aggressive soft tissue sarcoma of endothelial cell origin. Cutaneous angiosarcoma (CA) accounts for 60% of cases of angiosarcoma. These include primary CA and secondary CA, caused by previous irradiation, chronic lymphoedema and pre-existing vascular malformation. About 50% of cutaneous angiosarcoma affects the head and neck region in elderly men, in particularly the scalp area. [1] Overall, sarcomas occur uncommonly in the head and neck, constituting less than 1% of all head and neck malignancies. [2] The prognosis for patients with angiosarcoma of the head and neck remains dismal, with a reported 5 years survival rate of nearly 10%. [3, 4, 5, 6]
CASE REPORT A 65 years old, male, presented to outdoor patient department of dermatology with 4 months history of indurated swelling over the left infra-orbital area. On examination, it was extending over the forehead, mid scalp and the other side of face. The scalp lesions began as small nodule which enlarged and increased in numbers gradually. There was no preceding trauma though the nodules bleed easily. The nodules ranged from 0.5 cm to 1 cm in diameter. Multiple nodules are ulcerated and bluish discoloration of skin was also present. No lymph node was palpable. The routine haematological investigations, renal function tests, liver function tests and creatine kinase were normal. X-ray sinus showed no evidence of sinusitis. Skin biopsy (punch by 5 mm) from temporal region was taken and sent for histo-pathological examination.
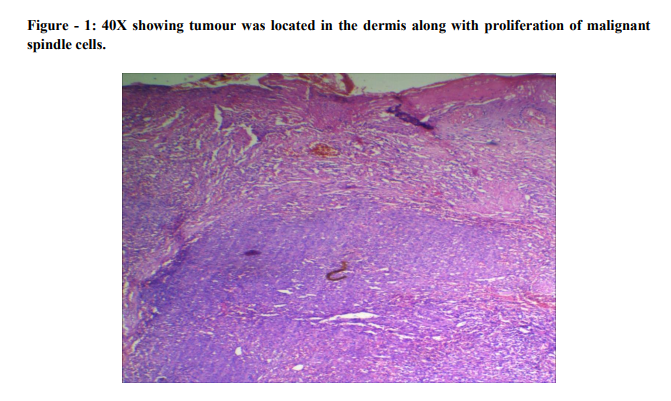
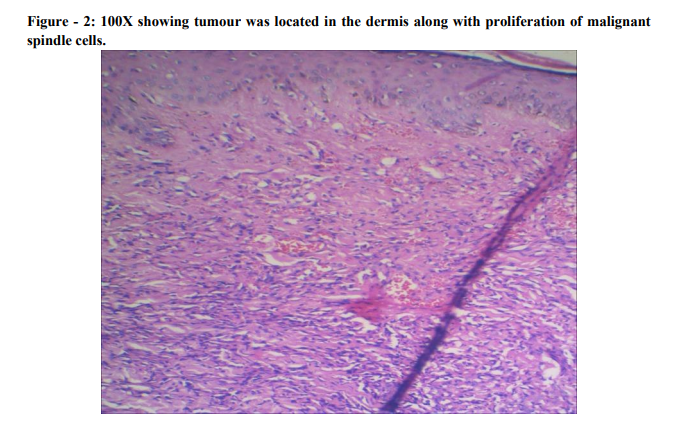
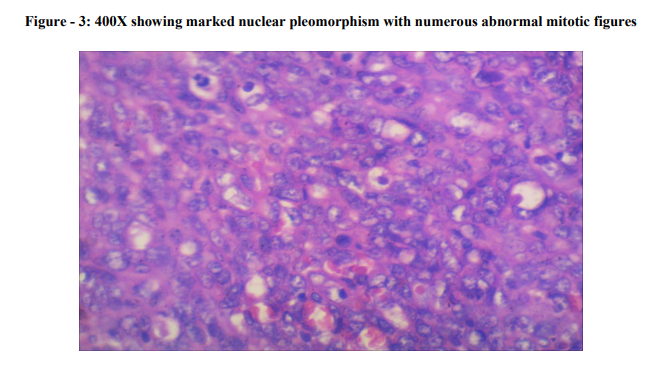
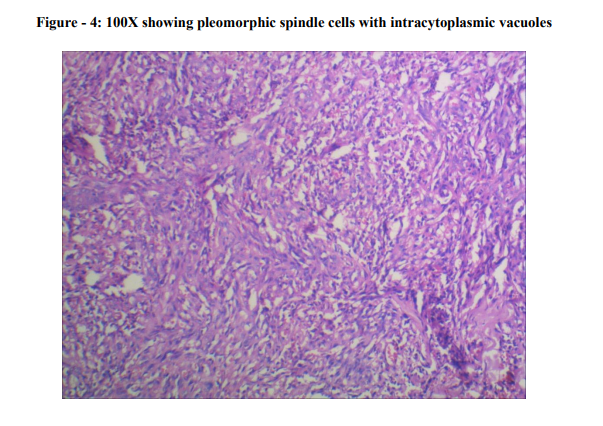
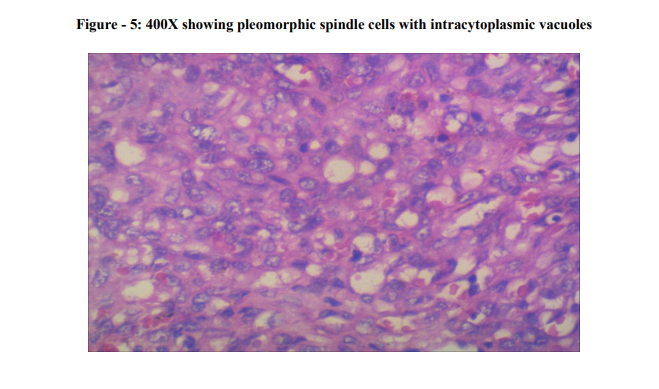
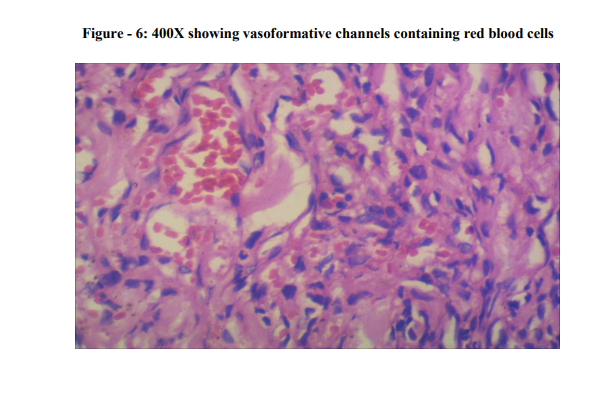
Microscopically, the tumour was located in the dermis. The tumour consisted of proliferation of malignant spindle cells without apparent differentiation. The tumour cells were seen to invade into the surrounding dermis and subcutaneous tissue (Photo - 1 and 2). Many mitotic figures were scattered (Photo - 3), and there were few areas of necrosis. Intracytoplasmic vesicles were recognized in several areas (Photo - 4 and 5). A few abortive vasoformative channels are also recognized. The presence of red blood cells in the vasoformative channels suggested the tumour was not a lymphatic tumour but a vascular tumour (Photo - 6). Hence, the diagnosis of cutaneous angiosarcoma was established.
DISCUSSION Cutaneous angiosarcoma (CA) was first described in 1945 by Caro and Stubenrauch. [7] In 1948, Stewart and Treves described association between angiosarcoma and postmastectomy lymphedema, the Stewart-Treves syndrome. [8] Wilson-Jones described cutaneous angiosarcoma primarily affects scalp and face of elderly in 1964. [9] CA is a rare neoplasm, accounting for <1% of all sarcomas. It commonly affects the head and neck regions (27.0%), followed by the breast (19.7%), extremities (15.3%), trunk (9.5%), liver (6.0%), heart (4.7%), bone (3.6%) and spleen (2.6%). [10] CA classify into primary and secondary. Primary CA is the more common form. In patients with primary CA, there has been no previous skin irradiation and lymphedema when compare to patients with secondary CA, which usually has either predisposing factor. Cutaneous form of angiosarcoma initially presents as a bruise or a raised purplish-red papule on the central face, forehead or scalp.
Facial swelling and oedema may be present. Increasing tumour size, tissue infiltration, oedema, tumour fungation, ulceration, haemorrhage can also develop. In lymphedema associated angiosarcoma (Stewart Treves syndrome), firm coalescing violaceous nodules or indurated plaque on a background of nonpitting oedema is the usual presentation. [11] Secondary CA is well known to affect the area of skin with chronic lymphedema and radio dermatitis. There is a male preponderance in primary CA, male to female ratio of 2-3:1. [12, 13] The histopathology of angiosarcoma depends on the areas of tumour differentiation. [14] In well differentiated areas, there is an anastomosing network of sinusoidal vessels lined by a single layer of endothelial cells of slight to moderate nuclear atypia. These vascular sinusoids split apart collagen bundles and groups of adipose cells. In less well differentiated areas, endothelial cells with more pronounced nuclear pleomorphism and mitotic activity pile up forming papillary projections. In poorly differentiated areas, the luminal formation is nonapparent. There is high mitotic activity mimicking high grade sarcoma or melanoma. Areas of haemorrhage or necrosis can occur.
Endothelial markers (von Willebrand factor, CD34, CD31, Ulex europaeus agglutinin 1, vascular endothelial growth factor (VEGF)) are expressed in angiosarcoma. [10] The absence of S100, human melanoma black 45, and melanoma antigen help to distinguish it from melanoma. The pathological diagnosis of angiosarcoma is easy when tumour cells form vascular channels. The diagnosis of angiosarcoma is very difficult if the apparent vascular channels are not recognized. Presence of foci of intra-cytoplasmic vacuoles is not pathognomonic for angiosarcoma, but they are suggestive for angiosarcoma. [15] The intra-cytoplasmic vacuoles may be the only clue of poorly differentiated angiosarcoma. In our study, we could suspect angiosarcoma because of the presence of these structures, but other sarcomas were also possible. Differential diagnosis includes exuberant granulation tissue, malignant vascular tumours, such as epithelioid haemangioendothelioma, perivascular myoid tumour (malignant glomus tumour and malignant myopericytoma), perivascular epithelioid tumour (malignant pecoma), kaposi's sarcoma, spindle cell carcinoma, intravascular endothelial hyperplasia, epithelioid angiosarcoma.
The present tumour was different from exuberant granulation tissue, because the present tumour showed malignant nature on Haematoxylin and Eosin (HE) findings. The current tumour was different from epithelioid hemangioendothelioma which shows more little atypia, no vasoformative channels, and collagenisation. The present case was histologically different from malignant glomus tumour, malignant myopericytoma, pecoma, kaposi's sarcoma, spindle cell carcinoma, intravascular endothelial hyperplasia and epithelioid angiosarcoma.
The overall 5 years survival is 10-35% [10] and the median survival of localised disease is 7 months. [16] Angiosarcoma has a tendency for metastasis by lymphatic or haematogenous routes, and late local recurrence and metastasis after years of apparent remission and successful local control are well documented. [17, 18] Local spread, recurrence and metastasis to regional lymph nodes and lungs are common. [16] Significant predictors of poor survival include: (1) tumour diameter >5 cm, (2) depth of invasion >3 mm, (3) mitotic figures >3/HPF, (4) positive surgical margin, (5) tumour recurrence and metastasis. [12] Management is by multidisciplinary approach. Combination of wide local excision, radiotherapy and chemotherapy had been adopted in various institutions.
CONCLUSION The rarity of primary angiosarcoma, unusual presentation of our case and possible diagnostic pitfalls associated with poor prognosis emphasizes the need for systemic presentation of these tumours in order to help pathologists and clinicians for early and accurate diagnosis with proper management. The aggressive nature of this disease demands careful follow-up of the patient and due to rarity of the condition, the appropriate strategies of treatment remain to be determined.
ACKNOWLEDGEMENT Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to authors / editors / publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed.
References:
1. Morgan MB, Swann M, Somach S, et al., Cutaneous angiosarcoma: a case series with prognostic correlation, Journal of American Academy of Dermatology, 2004; 50: 867- 874.
2. Figueiredo MT, Marques LA, Campos-Filho N., Soft tissue sarcomas of the head and neck in adults and children: Experience at a single institution and a review of the literature, International Journal of Cancer, 1988; 41: 198–200.
3. Holden CA, Spittle MF, Jones EW, Angiosarcoma of the face and scalp, prognosis and treatment, Cancer, 1987; 59: 1046–1057.
4. Liu AC, Kapp DS, Egbert B, et al., Angiosarcoma of the face and scalp, Annals of Plastic Surgery, 1990; 24: 68 –74.
5. Hodgkinson DJ, Soule EH, Woods JE, Cutaneous angiosarcoma of the head and neck, Cancer, 1979; 44: 1106–1113.
6. Maddox JC, Evans HL, Angiosarcoma of skin and soft tissue: a study of 44 cases, Cancer, 1981; 48: 1907–1921.
7. Caro M, Stubenrauch C., Hemangioendothelioma of the skin, Archieves of Dermatology and Syphilis, 1945; 51: 295-301.
8. Stewart FW, Treves N., Lymphangiosarcoma in postmastectomy lymphedema, Cancer, 1948; 1: 64-81.
9. Wilson-Jones E., Malignant angioendothelioma of the skin, British Journal of Dermatology, 1964; 76: 21-39.
10. Young RJ, Brown NJ, Reed MW, et al., Angiosarcoma, The Lancet Oncology, 2010; 11: 983-991.
11. North PE, Kincannon J., Vascular neoplasms and neoplastic like proliferations, Bolognia JL, Jorizzo JL, Rapini RP’s Dermatology, 2nd edition, Elsevier; United States, 2008, p.1788-1790.
12. Morgan MB, Swann M, Somach S, et al., Cutaneous angiosarcoma: A case series with prognostic correlation, Journal of American Academy of Dermatology, 2004; 50: 867- 874.
13. Holden CA, Spittle MF, Jones EW., Angiosarcoma of the face and scalp, prognosis and treatment, Cancer, 1987; 59: 1046-1057.
14. North PE, Kincannon J., Vascular neoplasms and neoplastic like proliferations, Bolognia JL, Jorizzo JL, Rapini RP’s Dermatology, 2nd edition, Elsevier, United States; 2008, p.1788-1790.
15. Fisher C., Soft tissue tumour, LeBoit PM, Burg G, Weedon D, Sarasin A’s World Health Organization Classification of Tumours, Pathology and genetics of skin tumours, Lyon: IARC Press; 2006, p. 230– 262.
16. Abraham JA, Hornicek FJ, Kaufman AM, et al., Treatment and outcome of 82 patients with angiosarcoma, Annals of Surgical Oncology, 2007; 14: 1953-1967.
17. Rosai J, Sumner HW, Kostianovosky M, et al., Angiosarcoma of the skin: A clinicopathologic and fine structural study, Human Pathology, 1976; 7: 83–109.
18. Liu AC, Kapp DS, Egbert B, et al., Angiosarcoma of the face and scalp, Annals of Plastic Surgery, 1990; 24: 68 –74.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





