IJCRR - 6(9), May, 2014
Pages: 145-155
Print Article
Download XML Download PDF
CURRENT TREATMENT PARADIGMS FOR RETINOBLASTOMA
Author: Shanti Pandey, Kavita Pandey, Vijay Joshi, Swati Gupta, Kalpana
Category: Healthcare
Abstract:Retinoblastoma is a primary malignant neoplasm of the retina that arises from immature retinal cells
which affects infants and children and it is the most common primary tumor of the eye in children.
Retinoblastoma detected on time can lead the child to see and moreover can save his life.
Retinoblastoma in later stages can endanger life. It becomes essential to know most about this life
threating as well as sight threating condition. Let these children live and enjoy the color of life.
Keywords: Retinoblastoma, chemotherapy, tumor, eye
Full Text:
INTRODUCTION
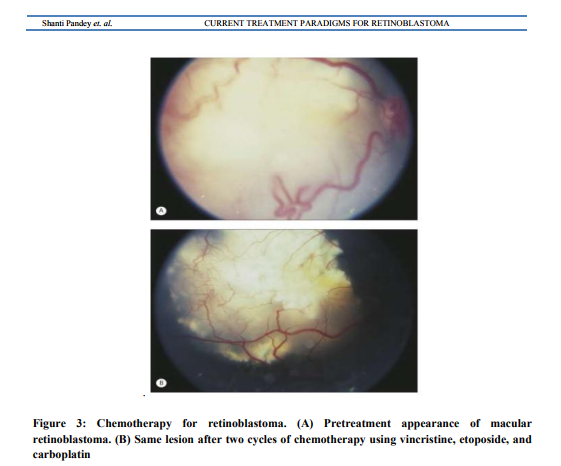
Retinoblastoma is a primary malignant neoplasm of the retina that arises from immature retinal cells which affects infants and children and it is the most common primary tumor of theeye in children.1 Out of newly diagnosed cases of retinoblastoma only 6% are familial while 94% are sporadic. Bilateral retinoblastoma are germline in all cases .Only 15% of unilateral retinoblastoma are caused by germline mutation, remaining 85% are sporadic. 2In 1971,Knudson proposed twohit hypothesis suggesting that in hereditary cases,germline mutations are present in all cells and somatic mutation occur only in retinal cells so they predisposed to nonocular carcinomas whereas in unilateral sporadic cases both hits occur during development of retina so no risk of secondary malignancy.3 In 1800s enucleation was the primary therapy to save life of the child. Then in early 1900s, external beam radiotherapy salvaged the life of child at the cost ofpoor vision and phthisis bulbi, secondary malignancy and orbital bone growth retardation. Refinements in radiotherapy including plaque beam therapy,laser therapy, thermotherapy, cryotherapy and latest being,periocular carboplatin injections has been done. In 1996, Kingston et al4 showed that ocular salvage rate improved to 90% if chemoreduction therapy is given prior to radiotherapyfor intraocular retinoblastoma. In a study on 158 eyes treated with vincristine, etoposide and carboplatin regimen given in six cycles ,allretinoblastoma,vitreous seeds and subretinal seeds initially regressed butatleast one vitreous seed recurrence was seen in 50% cases and at least one subretinal seed recurrence in 62% cases at 5 years follow up.5 Recent concept is of synergistic therapy (chemothermotherapy or chemocryotherapy) applying chemoreduction and focal tumor consolidation consecutively on tumor and seeds. Shields et al6 showed the recurrence in 18% cases treated with chemoreduction and thermotherapy compared with recurrences in 45% cases treated with chemotherapy alone in 7 year follow up in 457 patients of retinoblastoma. Studies showed similar effect on macular retinoblastoma.7 Various studies on high risk retinoblastomas showed that high dose chemotherapy in
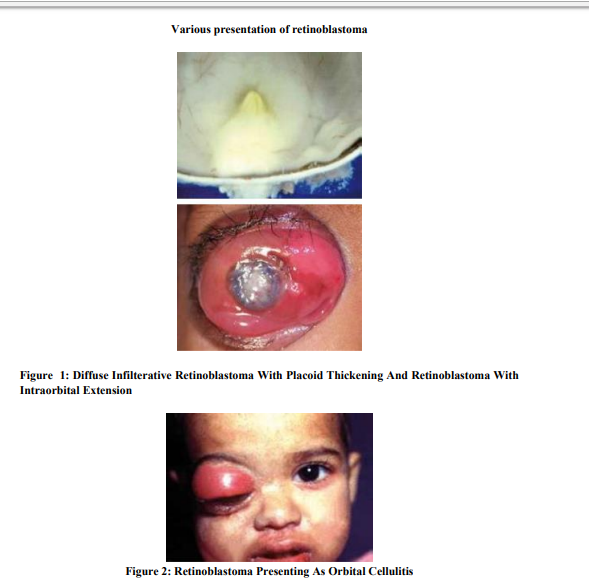
Diagnosis of retinoblastoma
A thorough clinical evaluation with indirect ophthalmoscopy and ultrasonography B-scan, can establish the diagnosis of retinoblastoma.14 USG B-scan shows typical intra-lesional calcification of retinoblastoma. Computed tomography and magnetic resonance imaging are indicated when extraocular or intracranial spread of tumor is suspected.14Retcam is a wide angle fundus camerawhich helps in documenting retinoblastoma and monitoring of tumor regression on follow up.
Classification of retinoblastoma
The Reese Ellworth classification was introduced to prognosticate patients treated with methods other than enucleation mainly external beam radiotherapy.9 Group I a) solitary tumor, < 4 disc diameters in size, at or behind the equator b) multiple tumor, none > 4 disc diameters in size, at or behind the equator Group II a.) solitary tumor, 4–10 disc diameters in size, at or behind the equator b) multiple tumor, 4–10 disc diameters in size, behind the equator Group III a) any lesion anterior to the equator b) solitary tumor >10 disc diameters behind the equator Group IV a)multiple tumors, some >10 disk diameters b) any lesion extending anteriorly to the oraserrata Group V a)massive tumor involving more than half of retina b) vitreousseedings International retinoblastoma staging systemis based on collective information gathered by the clinical evaluation, imaging, systemic survey and histopathology.10 Stage 0: No enucleation(one or both eyes may have intraocular disease) Stage I: Eye enucleated, completely resected histologically Stage II: Eye enucleated with microscopic residual tumor Stage III: Regional extension a) Overt orbital disease b) Preauricular or cervical lymph node extension Stage IV: Metastatic disease a) Hematogenous metastasis 1. single lesion 2. multiple lesions b) CNS extension: 1. Prechiasmatic lesion 2. CNS mass 3. Leptomeningeal disease Management of retinoblastoma The management of retinoblastoma is a multidisciplinary approach, depends on the stage of the disease and it is highly individualized. There are several methods to manage intraocular retinoblastoma namely- focal methods like cryotherapy, laser photocoagulation, transpupillary thermotherapy and plaque brachytherapy; local methods like enucleation and external beam radiotherapy and systemic therapy like chemotherapy Chemotherapy Chemotherapy is currently the primary therapeutic option in most children with bilateral retinoblastoma11and in some children with unilateral disease when the affected eye is believed to be salvageable. Chemoreduction refers to a process involving reduction in tumor volume after chemotherapy.It is not effective alone but in adjunct with focal therapy it can avoid enucleation or external beam radiotherapy without significant toxicity.3,12,13 The most common chemotherapeutic regimen in use around the world today consists of a combination of carboplatin, etoposide or a related drug, and vincristine (CEV regimen) for 6 cycles. In some centers, cyclosporine is added to this regimen to reduce the multidrug resistance that occurs in many retinoblastomas.15 Several promising new drugs and alternative regimens are currently being evaluated.Thiotepaused in the treatment of ovarian and breast cancer, penetrates well intothe brain, and has been considered for HDC. The investigators observed considerable, but manageable, toxicity with high-dose thiotepa. A response of retinoblastoma to conventional dose thiotepa is documented in one case.16For all these reasons, Kremens et al 17 decided to include thiotepa into the HDC regimen for children with high-risk RB.Thus High Dose Chemotherapy withthiotepa, etoposideand carboplatin may represent a curative option for children with extrabulbar or disseminated retinoblastoma. Standard chemotherapy is given in 6 cycles; after first 2 cycles, local therapy in form of cryotherapy, thermotherapy or plaque radiation may be given to completely regressed tumors.18Chemoreduction therapy is most successful for tumors without intravitreal and subretinal seedings5,19Risk factors for recurrence of tumor ,subretinal seed and vitreous seed recurrence include5,19 a. Tumor with subretinal seeding at initial presentation b. Younger patients with large bulky tumor. In cases of recurrence ,external beam radiotherapy and/or enucleation is required. A study revealed that non-caucasian race, male sex and Rees Ellsworth group V had higher rate of tumor recurrence.19 Adverse effects of chemotherapy include myelosupression, febrile episodes ,neurotoxicities and non specific gastrointestinal toxicities Matsubara Het al20used high-dose chemotherapy (HDC) with autologous stem cell transplantation (SCT) in patients with metastatic retinoblastomawithout CNS involvement. Melphalan was a key drug, and was administered in combination with other agents such as cisplatin, cyclophosphamide, carboplatin or thiotepa. Similarly Dunkel IJ et al21 suggested an intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. They concluded that intensive multimodality therapy including highdose chemotherapy(carboplatin and thiotepa alone or with etoposide or topotecan) with autologous hematopoietic stem cell rescue was curative for the majority of patients with stage 4a metastatic retinoblastoma. The contribution of external beam radiation therapy is unclear. They also suggestedsimilar therapy for stage 4B retinoblastoma(central nervous system metastatic disease)22and concludedthatintensive multimodality therapy may be beneficial for such patients but longer follow-up is required to determine whether it has been curative. They also described a series of 13 patients with trilateral retinoblastoma treated with similar intensive chemotherapy, and concluded that intensive chemotherapy is potentially curative for some patients with trilateral retinoblastoma, especially those withlocalized (M-0) disease.23 Periocular Chemotherapy Periocular(subtenon) carboplatin injections are currently being evaluated as an adjunct to intravenous chemotherapy in management of Rees Ellsworth Group VB retinoblastoma with vitreous seeds. Honavar et al 24did study which showed that periocular chemotherapy can achieve 70% eye salvage in patients with retinoblastoma with diffuse vitreous seeds.
Enucleation
Enucleation still remains important therapeutic option for unilateral advanced intraocular retinoblastoma and some cases of bilateral faradvanced disease not amenable to any eyepreserving therapy.Primary enucleation remains treatment of choice for advanced intraocular retinoblastoma with neovascularisation of iris,secondary glaucoma, anterior chamber tumor invasion,tumors occupying >75% of vitreous volume,necrotic tumors with secondary orbital inflammation and tumors with hyphema or vitreous hemorrhage where tumor characteristics cannot be recognized especially when only one eye is involved.14

During enucleation10- 15mm long section of the optic nerve should be removed as the principal route of exit of tumor cells from the eye is along the optic nerve for which traction sutures applied over recti muscles and a 15 degree blunt tipped and angled tenotomy scissors or even straight scissors may be used. Pathological studies have shown enucleation to be curative if >5mm of optic nerve is sacrificed. 26 Necrotic tumor with aseptic orbital cellulitis and phthisis bulbi should be imaged before enucleation to rule out extraocular extension. Enucleation should be done when inflammation has subsided25 with brief course of preoperative oral and topical steroids.25Phthisis bulbi usually follows spontaneous tumor necrosis and an episode of aseptic orbital cellulitis. Enucleation in these cases is often complicated by intraoperative bleeding and excessive periorbital fibrosis.25 Insertion of an orbital implant at the time of enucleation appears to be appropriate except when there is a strong likelihood of residual tumor in the orbit which may require orbital radiation therapy. Biointegrated implants are avoided if adjunctive postoperative radiotherapy is necessary because implant vascularity is compromised by radiotherapy which increases risk of implant exposure25. Myoconjunctival technique and custom ocular prosthesis have optimized prosthetismotility.and static cosmesis. External Beam Radiation Therapy Now a days external beam radiation therapy is indicated when focal therapy or chemotherapy fails to control tumor or very rarely when chemotherapy is contraindicated14.It is also applicable to eyes containing one or more tumors that involve the optic disc and eyes that show diffuse intravitreal or subretinal seeding.Standard target doses of radiation to the eye and orbit are in the range of 40–50 Gy given in multiple fractions of 150–200 cGy over 4–5 weeks. External beam radiation therapy results in two main patterns of regression. In the first pattern (type I), the tumor regresses to an almost exclusively calcific, avascular residual mound. In the second pattern (type II), the tumor regresses without prominent calcification but with a gray-tan fish-flesh appearance. The dilated retinal vessels usually become markedly attenuated with both regression patterns. In type III regression, a combination of both regression patterns occurs. Major complications of external beam radiation therapy are stunting of orbital growth leading to cosmetic facial deformity28, dry eye, radiation cataract27, radiation retinopathy and optic neuropathy. Cataract typically begins as posterior subcapsular clouding and usually does not form for at least 6 months after radiation therapy and is often delayed for as much as 1–1.5 years after treatment. External beam radiation therapy also increases the risk of nonretinoblastoma malignancies in the field of treatment in survivors of germinal retinoblastoma29,30 by 30%compared to less than 6% chance in those who do not receive external beam radiotherapy. This effect also appears most pronounced in children irradiated before the age of 1 year.31
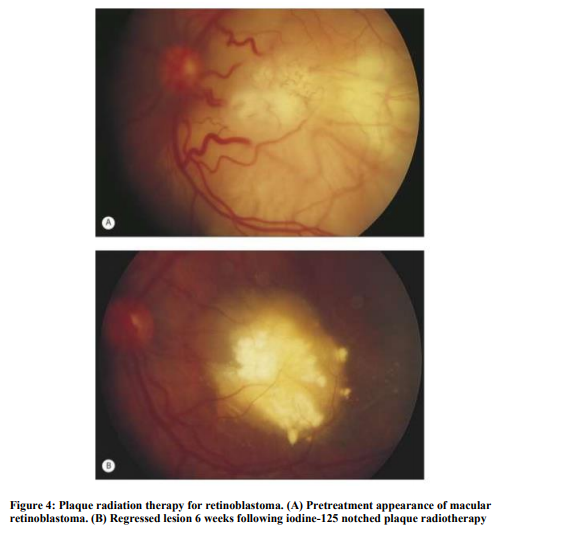
Plaque Radiation Therapy
In some children who have relatively large but localized retinoblastomas (less than 16mm in basal diameter and less than 8 mm in thickness), that does not involve optic disc or maculaeven in the presence of limited localized vitreous seeding, plaque radiation therapy may be employed successfully.32,33 Plaque radiation therapy entails surgical implantation of a radioactive device (eye plaque containing radioisotope iodine-125 and ruthenium106) of appropriate size and strength on the sclera overlying the intraocular tumor, leaving the plaque in place for a sufficient period of time (usually 2–5 days) to provide a predetermined radiation dose to the apex of the tumor, and subsequent surgical removal of the plaque. Currently it is performed as primary management only when chemotherapy is contraindicated. It is most usefull as secondary management in tumors which fail to respond to either chemotherapy or external beam radiotherapy or in cases of tumor recurrences. The advantages of plaque radiation therapy are focal delivery of radiation with minimal damage to surrounding normal structures, minimal periorbital tissue damage, absence of cosmetic deformity.
Laser photocoagulation
In photocoagulation, an argon green laser is employed using an indirect ophthalmoscope delivery system to delimit the tumor and coagulate the blood supply with two rows of overlapping laser burns.Laser photocoagulation is useful for small tumor 4mm in basal diameter and 2mm in thickness.14 Complications are transient serous retinal detachment, retinal vascular occlusion, retinal hole, retinal traction and preretinal fibrosis.Now with the advent of thermotherapy laser photocoagulation is less commonly used. Infact it is contraindicated when patient is on active chemoreduction protocol.14 Thermotherapy In transpupillary thermotherapy (TTT),34 an infrared laser beam (wavelength 810 nm) is directed at the retinal tumor using an operating microscope or indirect ophthalmoscope delivery systemto induce tissue necrosis.35by achieving a temperature of 40-60 degree celsius within the tumor with sparing of retinal vesselsuntil it appears homogeneously dull white.Large spot sizes (generally 2–3mm in diameter) are used if the pupil can be dilated widely. Infrared radiation can also be given by transscleral route with a diopexy probe. Thermotherapy is most appropriate for small intraretinal(4mm in basal diameter and 2mm in thickness)extramacular and extrapapillary tumors in eyes with clear optic media.35.It is not applicable to tumor associated with intravitreal or subretinal tumor seeds or retinal detachment.Common complications of thermotherapy are focal iris atrophy, focal paraxial lens opacity, retinal traction and serous retinal detachment. Major application of thermotherapy is as an adjunct to chemotherapy. The heat amplifies cytotoxic effects of platinum analogues. This synergistic combination with chemoreduction therapy is known as chemothermotherapy.
Cryotherapy
Trans-scleral cryotherapy is an obliterative focal treatment method that destroys targeted intraocular tissues by means of freezing36using an insulated retinal cryoprobeand indirect ophthalmoscopy.Double freeze-thaw method or triple freezethaw method may be applied according to the size of the tumor. This treatment is most applicable to small to medium size retinal tumors with minimal or no intravitreal or subretinal seeds or associated retinal detachment when the tumors are located in theequatorial or pre-equatorial region of the fundus.36Complications include serous retinal detachment, rhegmatogenous retinal detachment and retinal tear. Cryotherapy administered 2-3 hrs. before chemotherapy increase the delivery of chemodrugsacroos the blood retinal barrier and thus have synergistic effects.14
High risk retinoblastoma
High risk factors predictive of systemic metastasis include anterior chamber seedings, iris infiltration, ciliary body infiltration, massive choroidal infiltration, retrolaminar optic nerve invasion, invasion of optic nerve up to transaction, scleral infiltration and extrascleral extension.37 Current protocol is to administer standard dose chemotherapy including carboplatin, vincristine and etoposide in six cycles following enucleation in high risk charactersticpatients . Those cases with invasion of optic nerve transaction, sclera infiltration or extrascleral extension are treated with 12 cycles of HDC, orbital external beam radiotherapy, and/or enucleation8
Orbital retinoblastoma
Orbital retinoblastoma refers to orbital extension of retinoblastoma in the form of sclera infiltration, extrascleral extension or optic nerve invasion .When orbital extension is present at the time of diagnosis of tumor, it refers to primary orbital retinoblastoma. Orbital recurrence after uneventful enucleationfor intraocular retinoblastoma is referred to as secondary retinoblastoma. Sometimes inadvertent perforation, fine needle aspiration biopsy or intraocular surgery is done in cases of unsuspected retinoblastoma, orbital extension and systemic dissemination occurs. This orbital extension is called as accidental orbital retinoblastoma. Previously unrecognized orbital extension discovered macroscopically or microscopically at time of enucleation is referred as overt or microscopic overt retinoblastoma.38 Technetium -99 bone scan and PET- computed tomography can help in early detection of these subclinical metastasis.
Honavar etal developed a multimodality treatment protocol for management of orbital retinoblastoma which begins with 3 cycles of high dose chemotherapy followed by enucleation or orbital exentration, orbital external beam radiotherapy and finally extended 12 cycles of standard dose chemotherapy. Early results of this treatment protocol are encouraging.39 Some cases of retinoblastoma require intraocular surgery like cataract surgery for radiation cataract, sclera buckling or pars planavitrectomy for retinal detachment or vitreous hemorrhage. Specific guidelines for intraocular surgery after treatment of retinoblastoma have been described40
Metastatic retinoblastoma
Metastasis in retinoblastoma, which commonly spreads to orbital and regional lymph nodes, CNS, bones and bone marrow, usually develops within first year of diagnosis and the chances are very less after 5 years of diagnosis.Until recently the prognosis of metastatic disease was very poor. Various studies using high dose chemotherapy with autologous stem cell transplantation produced promising results in the cure of metastatic retinoblastoma especially those in bone and bone marrow20,21,22,23. Still CNS metastasis have grave prognosis and longer follow up is required to confirm the results of these studies.
Genetic counseling
It is an important aspect in management of retinoblastoma. In patients with positive family history,40% of the siblings would be at risk of developing retinoblastoma and 40% of the offsprings may develop retinoblastoma. In unilateral non familial retinoblastoma,1% of siblings and 8% of offsprings would be affected. In cases of bilateral nonfamilial retinoblastoma,6% of siblings and 40% of offsprings are at risk of developing retinoblastoma2 .Various studies on mutational analysis of RB1 gene have shown that by identifying specific mutations ,screening tests can be designed which help in computing specific ante natal risks.41
CONCLUSION
Retinoblastoma is a primary malignant neoplasm of the retina that arises from immature retinal cells which affects infants and children and it is the most common primary tumor of the eye in children. Retinoblastoma detected on time can lead the child to see and moreover can save his life. Retinoblastoma in later stages can endanger life. It becomes essential to know most about this life threating as well as sight threating condition. Let these children live and enjoy the color of life.
References:
REFERENCES
1. Donaldson SS, Egbert PR, Lee W. In: Pizzo PA, PoplackDG(eds). Principles and Practice of Pediatric Oncology.JBLippincott: Philadelphia, 1993, pp 683–696.
2. SheildsJA,Sheilds CL. Management and prognosis of retinoblastoma.In intraocular tumors. A text and atlas Philadelphia:WB Saunders 1992;337-392.
3. KnudsonAG:Mutation and cancer:Statistical study of retinoblastoma.ProcNatlAcadSci,USA 1971:68:820-823.
4. Kingston JE, Hungerford JL, Madreparla SA, Plowman PN. Results of combined radiotherapy and chemotherapy for advanced intraocular retinoblastoma. Arch Ophthalmol 1996;114:1339-1343.
5. Shields CL, Honavar SG, Shields JA, Dimirci H, Medows AT, Naduvilath TJ. Factors predictive of recurrence of retinal tumors ,vitreous seed, and subretinal seeds following chemoreduction for retinoblastoma. Arch Ophthalmol 2002;120:460-464.
6. Shields CL, MashayekhiA ,Shelids JA, Cater J, Medows AT, Shields JA. Chemoreduction for retinoblastoma. Analysis of tumour control and risk of recurrence in 457 tumors.AM J Ophthalmol 2004;138:329-37.
7. Shields CL, MashayekhiA ,Shelil A, Ness S, Cater J, Medows AT, Shields JA.Macularretinoblastoma managed with chemoreduction. Analysis of tumor control with without thermotherapy in 68 tumors, Arch Ophthalmol 2005;123:765-73.
8. Honavar SG, Singh AD, Shields CL ,Medows AM, Dimirci H, Cater J, Shields JA . Postenucleation adjuvant therapy in high -risk retinoblastoma, Arch Ophthalmol 2002;120:923-931.
9. Ellsworth RM. The practical management of retinoblastoma. Trans Am OphthalmolSoc 1969:67:462-534.
10. Chantada G, Doz F, Antoneli CB ,et al A proposal for an international retinoblastoma staging system Pediatr Blood Cancer 2006:47;801-805.
11. Gombos D.S., Kelly A., Coen P.G., et al: Retinoblastoma treated with primary chemotherapy alone: the significance of tumor size, location, and age. Br J Ophthalmol 2002; 86:80-83.
12. Gallie BL, Budning A, Deboer G, Thiessen JJ, Koren G, Verjee Z, etal. Chemotherapy with focal therapy can cure intraocular retinoblastoma without radiotherapy. Arch Ophtalmol 1996;114:1321-8.
13. Shields CL, De Potter P, Himelstein BP ,Shields JA , Medows AT, Maris JM. Chemoreduction in the initial management of intraocular retinoblastoma.ArchOphthalmol 1996;114:1330-8.
14. Murthy R, Honavar SG, Naik MN, Reddy VA. Retinoblastoma. In: Dutta LC, ed. Modern Ophthalmology. New Delhi, India ,Jaypee Brothers;2004:849-859.
15. Chan H.S., DeBoer G., Thiessen J.J., et al: Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin Cancer Res. 1996; 2:1499-1508.
16. Heidemann RL, Cole DE, Balis F et al. Phase I and pharmacoqkinetic evaluation of thiotepa in the cerebrospinal fluid andplasma of pediatric patients: evidence for dosedependent plasma clearance of thiotepa. Cancer Res 1989; 49: 736–741.
17. B Kremens, R Wieland, H Reinhard, D Neubert, JD Beck, T Klingebiel, N Bornfeldand W Havers.High-dose chemotherapy with autologous stem cell rescue in childrenwith retinoblastoma. Bone Marrow Transplantation (2003);31: 281–284.
18. Shields C.L., Mashayekhi A., Cater J., et al: Chemoreduction forretinlblastoma: analysis of tumor control and risks for recurrence in 457 tumors. Trans Am Ophthalmol Soc. 2004; 102:35-44.
19. Shields CL, Honavar SG, Meadows AT, Shields JA, Demirci H, Singh A, Friedman DL, Naduvilath TJ. Chemoreduction plus focal therapy for retinoblastoma:factors predictive of need for treatment with external beam radiotherapy or enucleation. AmJ Ophthalmol.2002;133:657-64.
20. Matsubara H, Makimoto A, Higa T, Kawamoto H, Sakiyama S, Hosono A, Takayama J, Takaue Y, Murayama S, Sumi M, Kaneko A, Ohira M. A multidisciplinary treatment strategy thatincludes high-dose chemotherapy for metastatic retinoblastoma without CNS involvement.Bone Marrow Transplant. 2005 Apr;35(8):763-6.
21. Dunkel IJ, Khakoo Y, Kernan NA, Gershon T, Gilheeney S, Lyden DC, Wolden SL, Orjuela M, Gardner SL, Abramson DH. Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma Pediatr Blood Cancer. 2010 Jul 15;55(1):55-9.
22. Dunkel IJ, Chan HS, Jubran R, Chantada GL, Goldman S, Chintagumpala M, Khakoo Y, Abramson DH. High-dose chemotherapy with autologous hematopoietic stem cell rescue for stage 4B retinoblastoma.Pediatr Blood Cancer. 2010 Jul 15;55(1):149-52.
23. Dunkel IJ, Jubran RF, Gururangan S, Chantada GL, Finlay JL, Goldman S, Khakoo Y, O'Brien JM, Orjuela M, Rodriguez-Galindo C, Souweidane MM, Abramson DH. Trilateralretinoblastoma: potentially curable with intensive chemotherapy. Pediatr Blood Cancer. 2010 Mar;54(3):384-7.
24. Honavar SG, Shome D, Reddy VAP. Periocular carboplatin in the management of advanced intraocular retinoblastoma. Proceedings of the XII International congress of ocular Oncology,Vancouver,Canada,2005.
25. Honavar SG, Singh AD. Management of advanced retinoblastoma.OphthalmolClin North Am.2005;18:65-73.
26. Khelfaoui F., Validire P., Auperin A., et al: Histopathologic risk factors in retinoblastoma. A retrospective study of 172 patients treated in a single institution. Cancer 1996; 77:1206-1213.
27. Brooks H.L., Meyer D., Shields J.A., et al: Removal of radiation-induced cataracts in patients treated for retinoblastoma. Arch Ophthalmol 1990; 108:1701-1708.
28. Egbert P.R., Donaldson S.S., Moazed K., et al: Visual results and ocular complications following radiotherapy for retinoblastoma. Arch Ophthalmol 1978; 96:1826-1830.
29. Roarty J.D., McLean I.W., Zimmerman L.E.: Incidence of second neoplasms in patients with bilateral retinoblastoma. Ophthalmology 1988; 95:1583-1587.
30. Mohney B.G., Robertson D.M., Schomberg P.J., Hodge D.O.: Second nonocular tumors in survivors of heritable retinoblastomaand prior radiation therapy. Am J Ophthalmol 1998; 126:269-277.
31. Abramson DH, Frank CM. Second nonocular tumors in survivors of bilateral retinoblastoma; a possible age effect on radiation-related risk. Ophthalmology.1998;105: 573-580.
32. Shields C.L., Shields J.A., De Potter P., et al: Plaque radiotherapy in the management of retinoblastoma. Use as a primary and secondary treatment. Ophthalmology 1993; 100:216-224.
33. Schueler A.O., Fluhs D., Anastassiou G., et al: Beta-ray brachytherapy with (106) Ru plaques for retinoblastoma. Int J RadiatOncolBiol Phys. 2006; 65:1212-1221.
34. Abramson D.H., Schefler A.C.: Transpupillary thermotherapy as initial treatment for small intraocular retinoblastoma: technique and predictors of success. Ophthalmology 2004; 111:984-991.
35. Shields CL, Santos MC, Diniz W et al. Thermotherapy for retinoblastoma.ArchOphthalmol 1999; 117:885-893.
36. Shields J.A., Parsons H., Shields C.L., et al: The role of cryotherapy in the management of retinoblastoma. Am J Ophthalmol 1989; 108:260-264.
37. Singh AD, Shields CL, Shields JA. Prognostic factors in retinoblastoma.JPediatrOphthalmolStrab 2000;37:134-41.
38. Shields CL, Honavar S, Shields JA, Demirci H, Meadows AT. Vitrectomy in eyes with unsuspected retinoblastoma. Ophthalmology. 2000;107: 2250-5.
39. Honavar SG, Shields CL, Shields JA, Demrici H, Naduvilath TJ. Intraocular surgery after treatment of retinoblastoma. Arch Ophthalmol.2001;119: 1613-21.
40. Honavar SG, Reddy VAP, Murthy R, Naik M, Vemuganti GK. Management of orbital retinoblastoma.XI International Congress of Ocular Oncology. Hyderabad, India, 2004.pp.51.
41. Kiran VS, Kannabiran C, ChakravarthiK, Vemuganti GK, Honavar SG. Mutational screening of the RB1gene in Indian patients with retinoblastoma reveals eight novel and several recurrent mutations. Hum Mutat.2003;22:339.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





