IJCRR - 6(12), June, 2014
Pages: 33-42
Date of Publication: 23-Jun-2014
Print Article
Download XML Download PDF
\"CYCLOOXYGENASE-2\" - A VIBRANT CHEMICAL MEDIATOR IN A PLETHORA OFORAL LESIONS
Author: Aditi Amit Byatnal, Subhalakshmi Sen, Amit Byatnal, Monica Charlotte Solomon
Category: Healthcare
Abstract:Cyclooxygenase (COX) is an enzyme that is responsible for formation of important biological mediators called prostanoids, including prostaglandins, prostacyclin and thromboxane. Constituting a key regulatory enzyme of the ecosanoid biosynthetic pathway, COX catalyzes the conversion of arachidonic acid to protaglandinG2 (PGG2) and PGH2 and subsequently to a variety of eicosanoids. COX-2 is one of the isoforms of COX, which was upheld mainly as an inflammatory, inducible enzyme, having role different than that of COX-1. However, more recent studies are beginning to reveal additional functions of COX-2, especially in oral lesions.Most commonly studied area of interest being oral squamous cell carcinoma, along with salivary gland tumors, reactive inflammatory lesions and so on. The objective of this review is to discuss the role of COX-2 in various oral lesions.
Keywords: Cyclooxygenase, prostanoids
Full Text:
INTRODUCTION
The fascinating ability to treat fever and inflammation dates back about 3500 (400 B.C.) years ago to a time when the Greek physician Hippocrates prescribed an extract from willow bark and leaves. Later in the 17th century, the active ingredient of willow bark salicin was identified in Europe. The Kolbe company in Germany started mass producing salicylic acid in 1860. Acetylsalicyclic acid 1 (aspirin) the more palatable form of salicyclic acid was introduced into the market by Bayer in 18991 . However, the mechanism of action of anti-inflammatory and analgesic agents such as aspirin and indomethacin 2 remained elusive until the early 1960?s. This all changed in the seventies, when John Vane discovered the mechanism of action of aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) thereby increasing our ability to develop novel anti-inflammatory therapies1 . The success of NSAIDs in treating various inflammatory conditions such as rheumatoid arthritis (RA) and osteoarthritis (OA) validated inhibition of the enzyme prostaglandin H synthase (PGHS) or cyclooxygenase (COX) as a highly suitable target in anti-inflammatory therapies1,2 . However, the gastrointestinal (GI) toxicities associated with widespread NSAID use proved to be a major drawback during long term therapy3 . COX, a central enzyme in the biosynthetic pathway to prostaglandins from arachidonic acid, is a protein that was purified more than 20 years ago and cloned in 1988. In the early 90?s, Needleman, Simmons and Herschman?s group reported the presence of an inducible isoform of the enzyme COX later identified as Cyclooxygenase-2 (COX-2)4,5. This discovery led to the hypothesis that anti-inflammatory prostaglandins (PGs) were produced through constitutive expression of Cyclooxygenase -1 (COX-)1, whereas the proinflammatory PGs were produced via induction of the COX-2 isoform6 .
DISCUSSION
COX-2 and inflammation
During the inflammatory process, the COX-1 micro RNA (mRNA) and protein activity do not change whereas a dramatic increase in COX-2 levels occurs leading to increased production of proinflammatoryPGs.The expression of COX-2 has been studied extensively in animal models of inflammation which provided strong evidence that induction of COX-2 enzyme is associated with inflammation. The COX-1 enzyme does not appear to be affected by the inflammatory process since similar levels of mRNA and protein are detected in both normal and inflamed tissue in animal models. PGs such as PGE2 and PGI2 produced via the COX-2 pathway magnify the degree of inflammation initiated by other mediators of inflammation such as histamine and bradykinin leading to increased vascular permeability and edema7 . COX-2 is not detectable in normal tissue but is detectable after induction by inflammatory stimuli. Selective COX-2 inhibitors exhibit good anti-inflammatory and analgesic activities in various animal models. In humans, COX-1 is found to be constitutively expressed in a wide range of tissues including the kidney, lung, stomach, small intestine and colon. Hence, COX-1 is considered a housekeeping enzyme responsible for maintaining basal prostaglandin levels, which is important for tissue homeostasis. In contrast, most tissues do not routinely express COX-2 constitutively. It is only in the central nervous system8 and seminal vesicles9where COX-2 has been demonstrated to be expressed normally. However, the stimulation of COX-2 in Src-transformed fibroblasts10 , endothelial cells and monocytes treated with the tumor promoter tetradecanoyl-phorbol-acetate or lipopolysaccharide create a notion that COX-2 is an inducible enzyme that produces prostaglandins during inflammatory and tumorigenic settings. Cyclooxygenase-2 and its function One of the first studies conducted after the discovery of two isoforms of COX was a screening of existing NSAIDs for those that had differential effects on inhibition of COX-1 vs. COX-2, and some were found to have a 20- to 70- fold selective preference11 . As a result, studies were done using differential inhibition of COX-1 or COX-2 activities to sort out the relative contributions of these isoforms under a variety of experimental conditions. COX-2 in Cancer Several population-based studies have detected a 40–50% decrease in relative risk for colorectal cancer in persons who regularly use aspirin and other NSAIDs12. Initial attempts to determine the molecular basis for these observations found that both human and animal colorectal tumors express high levels of COX-2, whereas the normal intestinal mucosa has low to undetectable COX-2 expression13. These findings led to the hypothesis that COX-2 may be playing a role in colon cancer growth and progression. Studies on cell culture models have shown that COX-2 expression contributes significantly to the tumorigenic potential of epithelial cells by increasing adhesion to extracellular matrix and making them resistant to apoptosis14. These phenotypic changes were shown to be reversible by treatment with a highly selective COX-2 inhibitor. Very recent work indicates that cyclooxygenase may play a vital role in the regulation of angiogenesis associated with neoplastic tumor cells15 . Hence, COX inhibitors may block the growth of blood vessels into developing tumors. COX-independent pathways also play an important role in the cancer chemopreventive properties of NSAIDs. Thus, it is understood that both COX-dependent and COXindependent pathways are involved in cancer chemoprevention
COX-2 in oral epithelial dysplasia and cancer
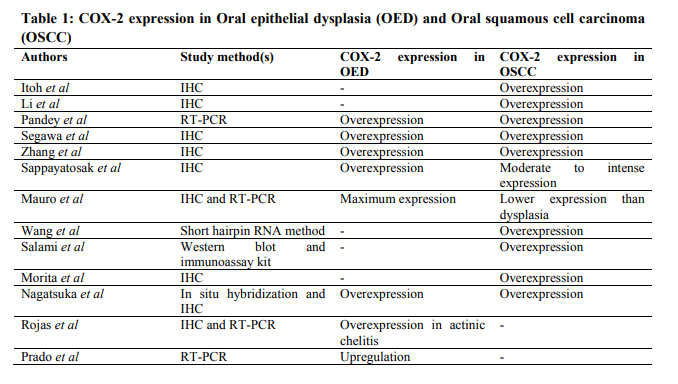
Expression of COX-2 has been evaluated in oral epithelial dysplasia and oral cancer. Results obtained are observed to be varying as depicted in ble 116-28 . It is well known that cancer cell development and survival is a multifactorial process, involving genetic mutation of normal cells as well as physiological changes within both cancer cells and also the body?s defense mechanisms29. Immune response to cancer cell development and progression is of particular importance as it might play a potential role in tumor formation. Unresolved immune responses, such as chronic inflammation, can promote the growth and progression of cancer. Within the immune system, cytotoxic CD8+ and CD4+ T cells, along with their characteristically produced cytokine interferon- γ (IFN-γ) function as the major anti-tumor immune effector cells, whereas tumor associated macrophages (TAM) or myeloidderived suppressive cells (MDSC) and their derived cytokines Interlukin-6, Tumor Necrosis Factor, Interlukin-1β and Interlukin-23 are generally recognized as dominant tumorpromoting forces. However, the roles played by CD4+, CD25+, Forkhead box (Fox) p3+ regulatory T lymphocytes and immunoregulatory cytokines such as Transforming Growth Factor -β in tumor development and survival remain elusive. These immune cells and the cellular factors produced from them, including both immunosuppressive and inflammatory cytokines, play dual roles in promoting or discouraging cancer development, and their ultimate role in cancer progression may rely heavily on the tumor microenvironment and the events leading to initial propagation of carcinogenesis30 . COX-2/PGE2 pathway has been demonstrated to influence every hallmark of cancer, including oral cancer. COX-2/PGE2 pathway has been suggested to play a role in suppression of apoptosis, via activation of the Ras-mitogen activated protein kinase (MAPK/ERK) pathway31. PGE2 has been reported to activate pro-survival pathways including PI3K/AKT pathway, protein kinase A signaling32 and activation of Epidermal Growth Factor (EGFR) signaling33. In addition to the apoptosis-suppressive effects of COX-2-derived PGE2, deregulated expression of COX-2 protein itself might also alter the susceptibility of cells to undergo apoptosis by reducing the cellular pool of its substrate arachidonic acid, which can stimulate apoptosis34. Aberrant activation of COX-2/PGE2 pathway might phenocopy activating mutations in the PI3K/ AKT and/or Ras-MAPK pathways, which could play an important role in promoting tumor progression. Indeed, deregulation of the COX-2/PGE2 pathway has been shown to behave in a similar manner to constitutively active Ras in murine intestinal adenomas, resulting in a positive feedback loop that boosts COX-2 expression and further stimulation of tumor growth35. COX2/PGE2 pathway prevents the receipt of antigrowth signals since over-expression of COX-2 has been reported to cause down-regulation of the TGFb type II receptor 14. The COX-2/PGE2 pathway, by enhancing cell survival and growth, serves to assist the cells for acquisition of further cellular alterations that contribute to immortalization and the progression towards the full malignant phenotype. Over-expression of COX-2 induces the production of angiogenic factors such as VEGF and basic fibroblast growth factor, which are instrumental in stimulating the formation of new blood vessels – a requirement for tumors should they wish to develop beyond a few millimeters in size36. The mechanism through which COX-2 might promote tumour vascularization is via the production of PGE2 and prostaglandin I2. These factors have been shown to participate in inducing endothelial cell dispersion and migration by integrin αVb3- mediated activation of the small guanosine 5?- triphosphatases Cdc42 and Rac37 . In vitro experiments have revealed that cells overexpressing COX-2 undergo phenotypic changes that could enhance their tumorigenic potential, such as exhibition of an increased adhesion to extracellular matrix proteins and resistance to apoptosis. It is believed that this proliferative activity of COX-2 primarily mediated by PGs14 . order to achieve metastases, cancer cells must exhibit a more motile, invasive phenotype, dissociate from neighboring cells within the tumour, invade through extracellular matrix components and intravasate into local blood or lymphatic channels38. Having made their escape from the primary tumour, cancer cells must then extravasate from the blood or lymphatics into the surrounding tissue in order to colonize distant sites. Several lines of evidence indicate that COX2 and the prostaglandins play important roles in aiding these processes – more specifically, PGE2 is thought to promote a more metastatic phenotype in cancer14 . Over-expression of COX-2 can modulate the adhesive properties of cancer cells and increase matrix metalloproteinase activity to promote invasion39. PGE2 promotes cytoskeletal reorganization and increases cancer cell migration and invasion via PI3K signaling. The stimulation of invasion and motility by PGE2 is dependent on the intracellular Src-mediated transactivation of EGFR40. Furthermore, hepatocyte growth factor signaling, which is classically associated with loss of cell-cell contact (or scattering) and invasive growth41, is also transactivated by PGE2 in an EGFR-dependent approach. COX-2, hepatocyte growth factor and β-catenin are co-expressed at the invasive front of tumor specimens42 , suggesting their interplay in tumorigenesis. COX2 has been identified as one of the four key „metastasis progression? genes, which collectively synergize to mediate both tumor development and metastasis to other organs43 . The contribution of COX-2 in carcinogenesis is due to its involvement in several key mechanisms including the conversion of pro-carcinogens to carcinogens as a consequence of arachidonic acid metabolism, stimulation of cell growth, inhibition of apoptosis through p53 suppression and bcl2 induction, stimulation of VEGF and angiogenesis, promotion of invasion and metastasis via matrix metalloproteinases induction and immunosuppression by IL-10 induction36,44. Cox-2 in potentially malignant oral lesions Along with numerous studies in conducted to evaluate the role of COX-2 in oral cancer, few studies have also been carried out to appraise if COX-2 plays a similar role in few of the lesions considered as potentially malignant oral lesions. As depicted in table 2 45-48, COX-2 has been shown to be overexpressed in oral submucous fibrosis (OSMF), oral lichen planus (OLP) and even in oral lichenoid reactions (OLR), as compared to the normal oral mucosa. It is well known that OLP is a chronic inflammatory disease for which the pathogenesis is not fully understood. OLP has autoimmune features and auto immunity has been suggested as a potential cause, whereas WHO has classified OLP as a premalignant condition. Association between chronic inflammation and cancer is known and chronic inflammation is one of the characteristics of OLP47. Increased expression of COX-2 suggests its definite role in OLP. COX-2 has been connected to both malignant development and autoimmunity but as malignant development of OLP is quite rare it was suggested that the increased levels of COX-2 support an autoimmune cause of the disease. As against, Lysitaet al48 suggest that sustained overexpression of COX-2 in the late stage of the disease could play a role in the malignant transformation of some OLP. In case of OSMF, very few studies have been conducted to assess the role of COX-2 in its pathogenesis. Aberrant and persistent tissue inflammations are believed to play an important role on the occurrence of tissue fibrosis. Tsai et al45 in their study demonstrated that COX-2 expression was significantly higher in OSMF specimens and expressed mainly by epithelial cells, endothelial cells, and cells with fibroblast morphology. Simultaneously they also observed the COX-2 expression in cells treated with arecoline and noticed that COX-2 expression was up-regulated as early as half an hour. This indicated that COX-2 expression is an early cellular response and regulated by arecoline at transcriptional level. Cox-2 in odontogenic cysts and tumors of the jaw Limitations in the number of studies related to expression of COX-2 in odontogenic cysts and tumors (table 3)49,50 makes it difficult to understand its exact role in pathogenesis of these lesions. Radicular cyst, being an inflammatory cyst, shows high expression of COX-2 in the lining epithelium, subepithelial fibroblasts, macrophages and endothelial cells, suggesting the role of COX-2 in their pathogenesis. KCOT is a benign neoplasm of odontogenic origin with an occasionally aggressive behavior leading to high recurrence rates. High COX-2 expression in the epithelial lining of KCOT the current knowledge of the role played by COX-2 in tumorigenesis further strengthen the current concept that the KCOT should be regarded as a neoplasm. Furthermore, the multitude of markers known to be overexpressed in KCOTs is suggestive of what could be called a 'network addiction' pattern, rather than a pathological mechanism dependant on a specific activated/suppressed gene, thus explaining its aggressive behavior50 . Cox-2 in salivary gland tumors Salivary gland tumors, most of which are rare benign tumors, represent a histologically heterogenous group with the greatest diversity of morphological and cellular features. Restricted number of studies has been conducted as to evaluate the role of COX-2 in few of the salivary gland tumors (table 4)51-55. Few studies showed no quantitative difference in COX-2 expression in different benign tumors51, while others exhibited considerable overexpression in the same52 . Warthin?s tumor showed COX-2 expression only in the epithelial component, with no expression in the lymphoid components54. These findings support the hypothesis that Warthin's tumors originate from heterotopic ductal epithelial cells of the parotid gland. However, the role of COX-2 expression in the pathogenesis of Warthin's tumors remains to be determined. Likewise, COX-2 expression has also been studied in malignant salivary gland tumors since overexpression of cyclooxygenase (COX)-2 in several human carcinomas suggests that COX-2 is related to carcinogenesis. However, the exact mechanism is still not understood55 . COX-2 in oral reactive lesions Most of the reactive lesions being inflammatory in process, have demonstrated COX-2 overexpression (table 5)56,57. Pulpal inflammation associated with dental caries56 andperiapical granuloma and periapicalcyst57have shown overexpression of COX-2. COX-2 -positive cells were detected in the epithelial cells and inflammatory cells. These studies suggest that COX-2 might play more important roles in the pathogenesis and development of periapical
CONCLUSION
Cyclooxygenase- 2 is chemical mediator that has a low to undetectable expression in normal tissues. It is a crucial element that functions through the production of prostaglandins. This molecule is particularly abundant in activated macrophages and cells at the site of inflammation. Its role in the disease process of various oral lesions is recognized. This mediator is a key factor in many regulatory pathways of the carcinogenic process. Understanding the role of cyclooxygenase -2 in the oral lesions can facilitate the incorporation of treatment protocols that target this molecule.
ACKNOWLEDGEMENT
Authors acknowledge the immense help received from the scholars whose articles have been cited and included in the references of this manuscript. The authors are also grateful to authors/ editor/ Publishers of all those articles, journal and books from where the literature for this article has been reviewed and discussed.

References:
REFERENCES
1. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biology 1991; 231: 232-235.
2. Ferreira SH, Moncada S, Vane JR. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nat New Biology 1971; 231: 237-239.
3. Tamblyn R, Berkson L, Dauphinee WD, Gayton D, Grad R, Huang A, Isaac L, McLeod P, Snell L. Unnecessary prescribing of NSAIDs and the management of NSAIDrelated gastropathy in medical practice. Ann Intern Med. 1997; 127(6): 429-38.
4. Masferrer JL, Zweifel BS, Manning P,T, Hauser SD, Leahy KM, Smith WG, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. ProcNatlAcadSci USA 1994; 91: 3228-3232.
5. Xie W, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogenresponsive gene encoding prostaglandin synthase is regulated by mRNA splicing. ProcNatlAcadSci USA. 1991; 88: 2692-2696.
6. Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1993;268(9):6610-4.
7. Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. ProcNatlAcadSci U S A. 1994;91(25):12013-7.
8. Svensson CI, Yaksh TL. The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Annu Rev PharmacolToxicol. 2002;42:553-83.
9. Kirschenbaum A, Liotta DR, Yao S, Liu XH, Klausner AP, Unger P, Shapiro E, Leav I, Levine AC. Immunohistochemical localization of cyclooxygenase-1 and cyclooxygenase-2 in the human foetal and adult male reproductive tracts. J ClinEndocrinolMetab. 2000;85(9):3436-41.
10. Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogenresponsive gene encoding prostaglandin synthase is regulated by mRNA splicing. ProcNatlAcadSci U S A. 1991;88(7):2692-6.
11. Spangler RS. Cyclooxygenase 1 and 2 in rheumatic disease: implications for nonsteroidal anti-inflammatory drug therapy. Semin Arthritis Rheum. 1996;26(1):435-46.
12. Giovannucci E, Rimm EB, Stampfer MJ, Colditz GA, Ascherio A, Willett WC. Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann Intern Med. 1994;121(4):241-6.
13. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Upregulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183-8.
14. Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83(3):493-501.
15. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93(5):705-16. Erratum in: Cell 1998;94(2):273.
16. Itoh S, Matsui K, Furuta I, Takano Y. Immunohistochemical study on overexpression of cyclooxygenase-2 in squamous cell carcinoma of the oral cavity: its importance as a prognostic predictor. Oral Oncol. 2003;39:829-35.
17. Li WZ, Ding YQ, Li ZG, Zhang JH. Expression of COX-2 and VEGF-C in squamous cell carcinoma of the tongue and its correlation to lymph node metastasis. Nan Fang Yi Ke Da XueXueBao. 2008;28(2):180- 3.
18. Pandey M, Prakash O, Santhi WS, Soumithran CS, Pillai RM. Overexpression of COX-2 gene in oral cancer is independent of stage of disease and degree of differentiation. Int J Oral Maxillofac Surg. 2008;37(4):379-83.
19. Segawa E, Sakurai K, Kishimoto H, Takaoka K, Noguchi K, Hashitani S, et al. Expression of cyclooxygenase-2 and DNA topoisomerase II alpha in precancerous and cancerous lesions of the oral mucosa. Oral Oncol. 2008;44(7):664-71.
20. Zhang S, Du Y, Tao J, Wu Y, Chen N. Expression of cytosolic phospholipase A2 and cyclooxygenase 2 and their significance in human oral mucosae, dysplasias and squamous cell carcinomas. ORL J OtorhinolaryngolRelat Spec. 2008;70(4):242- 8.
21. Sappayatosok K, Maneerat Y, Swasdison S, Viriyavejakul P, Dhanuthai K, Zwang J, et al. Expression of pro-inflammatory protein, iNOS, VEGF and COX-2 in oral squamous cell carcinoma (OSCC), relationship with angiogenesis and their clinico-pathological correlation. Med Oral Patol Oral Cir Bucal. 2009;14(7):E319-24.
22. Mauro A, Lipari L, Leone A, Tortorici S, Burruano F, Provenzano S, et al. Expression of cyclooxygenase-1 and cyclooxygenase-2 in normal and pathological human oral mucosa. Folia HistochemCytobiol. 2010;48(4):555-63.
23. Wang YH, Wu MW, Yang AK, Zhang WD, Sun J, Liu TR, et al. COX-2 Gene increases tongue cancer cell proliferation and invasion through VEGF-C pathway. Med Oncol. 2011;28Suppl 1:S360-6.
24. Salimi M, Esfahani M, Habibzadeh N, Aslani HR, Amanzadeh A, Esfandiary M, et al. Change in Nicotine-Induced VEGF, PGE2 AND COX-2 Expression Following COX Inhibition in Human Oral Squamous Cancer. J Environ PatholToxicolOncol. 2012;31(4):349- 56.
25. Morita Y, Hata K, Nakanishi M, Nishisho T, Yura Y, Yoneda T. Cyclooxygenase-2 promotes tumor lymphangiogenesis and lymph node metastasis in oral squamous cell carcinoma. Int J Oncol. 2012;41(3):885-92.
26. Nagatsuka H, Siar CH, Tsujigiwa H, Naomoto Y, Han PP, Gunduz M, et al. Heparanase and cyclooxygenase-2 gene and protein expressions during progression of oral epithelial dysplasia to carcinoma. Ann DiagnPathol. 2012;16(5):354-61.
27. Rojas IG, Martínez A, Brethauer U, Grez P, Yefi R, Luza S, et al. Actinic cheilitis: epithelial expression of COX-2 and its association with mast cell tryptase and PAR-2. Oral Oncol. 2009;45(3):284-90.
28. Prado SM, Cedrún JL, Rey RL, Villaamil VM, García AA, Ayerbes MV, et al. Evaluation of COX-2, EGFR, and p53 as biomarkers of nondysplastic oral leukoplakias. ExpMolPathol. 2010;89(2):197-203.
29. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140(6):883-99.
30. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. 2011;7(5):651- 8.
31. Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998;58(2):362-6.
32. Leone V, di Palma A, Ricchi P, Acquaviva F, Giannouli M, Di Prisco AM, et al. PGE2 inhibits apoptosis in human adenocarcinoma Caco-2 cell line through Ras-PI3K association and cAMP-dependent kinase A activation. Am J PhysiolGastrointest Liver Physiol. 2007;293(4):G673-81.
33. Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8(3):289-93.
34. Chan TA, Morin PJ, Vogelstein B, Kinzler KW. Mechanisms underlying nonsteroidalantiinflammatory drug-mediated apoptosis. ProcNatlAcadSci U S A. 1998;95(2):681-6.
35. Wang D, Buchanan FG, Wang H, Dey SK, DuBois RN. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade. Cancer Res. 2005;65(5):1822-9.
36. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353-64.
37. Dormond O, Foletti A, Paroz C, Rüegg C. NSAIDs inhibit alpha V beta 3 integrinmediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nat Med. 2001;7(9):1041-7.
38. Weinberg RA. Mechanisms of malignant progression. Carcinogenesis. 2008;29(6):1092-5.
39. Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. ProcNatlAcadSci U S A. 1997;94(7):3336-40.
40. Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003;278(37):35451-7.
41. Birchmeier C, Birchmeier W, Gherardi E, VandeWoude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915-25
42. Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signalling by cross-talk between two distinct growth factor receptors. FASEB J. 2003;17(12):1640-7.
43. Greenhough A, Smartt JM, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation of tumor microenvironment. Carcinogenesis. 2009;30(3):377-86.
44. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004;116 (2) 205-219.
45. Tsai CH, Chou MY, Chang YC. The upregulation of cyclooxygenase-2 expression in human buccal mucosal fibroblasts by arecoline: a possible role in the pathogenesis of oral submucous fibrosis. J Oral Pathol Med. 2003;32(3):146-53.
46. Danielsson K, Ebrahimi M, Wahlin YB, Nylander K, Boldrup L. Increased levels of COX-2 in oral lichen planus supports an autoimmune cause of the disease. J EurAcadDermatolVenereol. 2012;26(11):1415-9.
47. Cortés-Ramírez DA, Rodríguez-Tojo MJ, Gainza-Cirauqui ML, Martínez-Conde R, Aguirre-Urizar JM. Overexpression of cyclooxygenase-2 as a biomarker in different subtypes of the oral lichenoid disease. Oral Surg Oral Med Oral Pathol Oral RadiolEndod. 2010;110(6):738-43.
48. Lysitsa S, Samson J, Gerber-Wicht C, Lang U, Lombardi T. COX-2 expression in oral lichen planus. Dermatology. 2008;217(2):150-5.
49. Tsai CH, Huang FM, Yang LC, Chou MY, Chang YC. Immunohistochemical localization of cyclooxygenase-2 in radicular cysts. IntEndod J. 2002 Oct;35(10):854-8.
50. Mendes RA, Carvalho JF, van der Waal I. Potential relevance of cyclooxygenase-2 expression in keratocysticodontogenictumours – an immunohistochemical study. J Oral Pathol Med. 2011;40(6):497-503.
51. Lipari L, Mauro A, Gallina S, Tortorici S, Buscemi M, Tete S, Gerbino A. Expression of gelatinases (MMP-2, MMP-9) and cyclooxygenases (COX-1, COX-2) in some benign salivary gland tumors. Int J ImmunopatholPharmacol. 2012;25(1):107-15.
52. Cho NP, Han HS, Soh Y, Son HJ. Overexpression of cyclooxygenase-2 correlates with cytoplasmic HuR expression in salivary mucoepidermoid carcinoma but not in pleomorphic adenoma. J Oral Pathol Med. 2007;36(5):297-303.
53. Katori H, Nozawa A, Tsukuda M. Increased expression of cyclooxygenase-2 and Ki-67 are associated with malignant transformation of pleomorphic adenoma. AurisNasus Larynx. 2007;34(1):79-84.
54. Loy AH, Putti TC, Tan LK. Cyclooxygenase-2 expression in Warthin'stumour. J Laryngol Otol. 2005;119(7):515-8.
55. Sakurai K, Urade M, Noguchi K, Kishimoto H, Ishibashi M, Yasoshima H, Yamamoto T, Kubota A. Increased expression of cyclooxygenase-2 in human salivary gland tumors. Pathol Int. 2001;51(10):762-9
56. Lundy FT, About I, Curtis TM, McGahon MK, Linden GJ, Irwin CR, El Karim IA. PAR-2 regulates dental pulp inflammation associated with caries. J Dent Res. 2010;89(7):684-8.
57. Lin SK, Kok SH, Kuo MY, Wang TJ, Wang JT, Yeh FT, Hsiao M, Lan WH, Hong CY. Sequential expressions of MMP-1, TIMP-1, IL-6, and COX-2 genes in induced periapical lesions in rats. Eur J Oral Sci. 2002; 110(3):246-53.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





