IJCRR - 7(10), May, 2015
Pages: 11-13
Print Article
Download XML Download PDF
AN AUDIT OF RED CELL CONCENTRATES USAGE FOR THALASSEMIA PATIENTS VERSUS OTHER PATIENTS IN A TERTIARY CARE HOSPITAL AT RAJKOT
Author: Ghatna Kotadia, Amit Agravat, Gauravi Dhruva
Category: General Sciences
Abstract:Objective: To perform a retrospective audit of total Red Cell Concentrates (RCC) used for thalassemia patients in comparison with all other patients attended to Blood bank,P.D.U Medical College and Hospital,Rajkot. Materials and Methods: Each and every patient who attended blood bank are considered in the study with every episodes or blood transfusion in one year. Results: Total 4930 units of Red Cell Concentrates were issued to thalassemia patients in comparison with 9070 units of Redcell concentrates to other indoor patients.
Keywords: RCC for thalassemia patients
Full Text:
INTRODUCTION
Thalassemias are a group of hemolytic anemia which results from an inherited abnormality of globin production. Both alpha and beta thalassemia are the most common monogenetic disorder worldwide [1]. In India ,beta thalassemia is found throughout the country and an estimated 35 million people are carriers with a higher incidence in communities like Sindhi, Punjabi, Bengali, Gujrati, Parsi and Lohana mainly[2].Regular blood transfusion remains the mainstay of therapy in absence of curative treatment, such as stem cell transplantation. While stem cell transplantation is curative, issues of donor availability, access and cost make this option available to only a small fraction of individuals. Main objective of transfusion is to suppress ineffective erythropoiesis and to prevent anemia.
MATERIALS AND METHODS
Out of total 15671 patients attended the Blood Bank, P.D.U Medical College and Hospital, Rajkot Total 365 thalassemia patients were analyzed including all the episodes of blood transfusion from January 2014 to December 2014.On an average each patients of thlassemia major were transfused with one unit of red cell concentrate at an interval of every 15 to 20 days and thalassemia intermediate at a duration of 6 months. All thalassemia patients irrespective of their age were included in the study. In patients having history of transfusion related illness leucodepleted red cell concentrates were transfused.
RESULT
Out of total 14225 units of whole blood collected in blood bank, 9070 units of red cell concentrates were issued to indoor patients in comparison with 4930 units of red cell concentrates issued to thalassemia patients in a period of 1 month. It corresponds to 64.8% of total red cell concentrates used for indoor patients and 35.2% RCC usage for thalassemia patients.
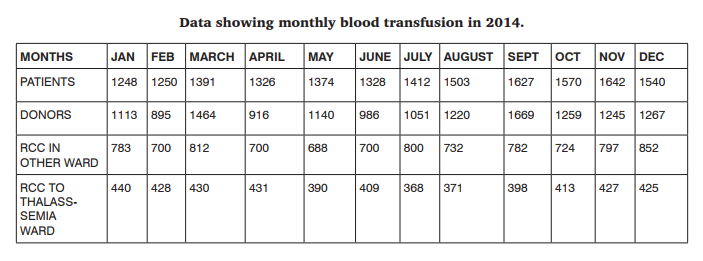
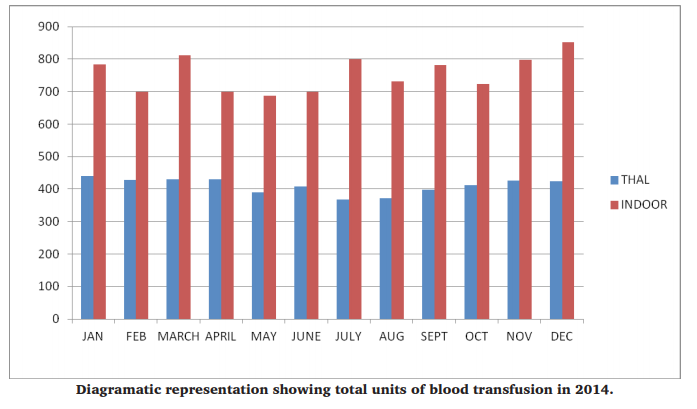
DISCUSSION
Thalassemia major is a complex disease which is common in Mediterranean regions particularly in India. Treatment for thalassemia has dramatically improved with patients living full life with carriers and children of their own. Unfortunately many patients die prematurely or develop complications. Prior to transfusion therapy it is necessary to confirm patient’s diagnosis by hemoglobin electrophoresis or by high performance liquid chromatography. Parents and siblings should be screened. Patients with thalassemia intermedia may have exaggerated anemia due to nutrition deficiency or any infection. It is important to elicit history for all these including any drugs, viral illness or environmental factors which can lower hemoglobin. Chronic transfusion prevents most of serious growth, skeletal and neurological complications of thalassemia major [3]. Irrational and long term use of transfusion in thalassemia patients without iron chelation can lead to hemosiderosis with resultant growth hormone and other endocrine deficiency leading to death [4]. Iron overload is the major cause of morbidity and mortality in thalassemia patients. The target is to keep the pre-transfusion hemoglobin level at 9 to 10 g/dl. The decision to start regular transfusion is clear when the initial hemoglobin level is well below 6g/dl. The decision to start transfusion is based on inability to compensate for low hemoglobin, or less commonly on increasing symptoms of ineffective erythropoiesis [5].The decision of chronic transfusion should not be based exclusively on presence of anemia. Febrile non-hemolytic transfusion reactions are frequent complication and can lead to additional costly medical interventions such as antipyretic and/or antibiotic usage as well as blood product wastage [6].The use of leukoreduced RBCs has been shown, in the general transfused population as well as in thalassemia patients specifically, to significantly reduce the incidence of febrile non-hemolytic transfusion reactions [7]. Annual transfusion volume exceeding 225 to 250 ml/kg per year with packed red blood cells indicate the presence of hypersplenism. Often hypersplenism develops because of low pre-transfusion hemoglobin. Increasing the pre-transfusion hemoglobin to between 9.5 to 10g/ dl may reverse hypersplenism.
CONCLUSION
Continous scrutinity regarding both the amount and duration of red cell concentrates transfusion must be done for thalassemia patients in order to prevent irrational use of red cell concentrates,iron toxicity and hypersplenism.
References:
1. Modell B, Darlison M.Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008;86:480-7.
2. Dr. Tejinder singh, Atlas and Text of HEEMATOLOGY,third edition,Hemolytic anaemia,p.g 133.
3. Piomelli S, Danoff SJ, Becker MH, et al. Prevention of bonemalformations and cardiomegaly in Cooley’s anemia byearly hypertransfusion regimen. Ann N Y Acad Sci 1969;165:427-36.
4. Schorr JB, Radel E. Transfusion therapy and its complications in patients with Cooley’s anemia. Ann N Y Acad Sci1964;119:703-8.
5. Wolman IJ. Transfusion therapy in Cooley’s anemia:growth and health as related to long-range hemoglobin levels. A progress report. Ann N Y Acad Sci 1964;119:736
6. Lane TA, Anderson KC, Goodnough LT, et al. Leukocytereduction in blood component therapy. Ann Intern Med1992;117:151-62.
7. Dzik S, Aubuchon J, Jeffries L, et al. Leukocyte reduction ofblood components: public policy and new technology. Transfus Med Rev 2000;14:34-52.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





