IJCRR - 14(6), March, 2022
Pages: 17-26
Date of Publication: 15-Mar-2022
Print Article
Download XML Download PDF
Whole Exome Sequencing Data Analysis for Detection of Breast Cancer Gene Variants and Pathway Study
Author: Dhanyakumar G, Maheswari L Patil
Category: Healthcare
Abstract:Introduction: Whole Exome Sequencing (WES) involves sequencing, analysis of protein-coding regions in genome. In present investigation, the potential gene variants were identified in human breast cancer genome using WES data analysis.
Materials and Method: The NGS data samples with accession numbers (SRR1274896_1, SRR1274896_2) and (SRR1275000_1, SRR1275000_2) were collected from ENA database. The quality of the samples was assessed by using FastQC tool and followed by aligning samples with reference genome sequence hg38 using the Bowtie2 tool. The results were retrieved in SAM format and converted to BAM format and then to sorted bam file using SAM tools, then duplicates were removed using Picard tool. Finally, Variant Calling format file was generated using BCF tools which projected the possible gene variants in the samples.
Results: The results showed variant types out of them MUC3A1 showed an average of 53 mutations, highlighting its importance as a potential gene variant observed in breast cancer. Out of nonsynonymous mutations of samples, common gene variants in samples that possess 5 and more mutations were selected. The study was carried out on pathway analysis, domain analysis, gene involvement in biological processes and gene function.
Conclusion: Majority of gene variants were involved in DNA Biosynthesis and Protein Biosynthesis and also resulted in tissue-specific location. The location of these genes showed mutated genes in cytoplasm and in nucleus indicating the impact of gene variation on intracellular process.
Keywords: Breast cancer, Mutations, MUC3A1, Next generation Sequencing (NGS), Whole exome sequencing, MUC16
Full Text:
INTRODUCTION
Breast cancer is the most commonly occurring cancer in female.1 Breast cancer is a complex disease with a variety of risk factors. The genetic factors, environmental factors and family history helps to determine the risk of breast cancer.2The hereditary breast cancers are caused by mutations in high-penetrance genes such as BRCA1 and BRCA2, p53, PTEN, ATM, NBS1 or LKB1.2, 3 There are low-penetrance genes such as MTHFR, CYP1A1, XRCC1 and XRCC3, ERCC4/XPF which are linked in an increase or decrease of breast cancer risk.2, 4, 5PI3K is considered to be the major signaling hub downstream of HER2 receptor tyrosine kinases, this pathway is commonly mutated in breast cancer as the mutational genes in this pathway occur about more than 70% of breast cancer.6 There exists an aggressive type of breast cancer subtype called Triple-negative breast cancer (TNBC), caused by lack of estrogen receptors (ERs), progesterone receptors (PgRs), and Erb-B2 Receptor Tyrosine Kinase 2gene.7 Due to the low incidence of this cancer type, still large scale clinical trials to be conducted in the future.8, 9
About 10% of breast cancer cases in women are linked to the hereditary due to mutations in genes.10The hereditary breast cancers are due to the mutations in BRCA1 and BRCA2.11The frequently mutated gene in breast cancer is Tp53.12Mutations in the PTEN gene is responsible for causing Cowden syndrome families also causes 25-50% lifetime breast cancer risk in women.13 Peutz-Jeghers syndrome can lead to breast cancer risk14which is caused by mutations in the LKB1 gene.15 Some of the breast cancer susceptibility genes other than BRCA1 and BRCA2 areTP53, PTEN, BARD1, BRIPI, MRE11A, NBN, RAD50, ATM, and CHEK2.16PIK3CA mutations occur during early mutational development of breast cancer and are common in ER+ (estrogen receptor) breast cancers.6 An ESR1 mutation is found in endocrine resistance in ER-positive breast cancer.17
Nowadays the cost of NGS has been reduced, the middle-class populations are able to afford NGS for preventing breast cancer.18 This investigation was focused on WES analysis of the breast cancer samples to identify the potential gene variants to predict the specific pathway, biological process and function that may be affected through the genetic mutations observed in gene variants in breast cancer conditions.
MATERIALS AND METHODS
Data set collection
The raw exome datasets of Breast cancer tissues SRR1274896 and SRR1275000 were downloaded from ENA database. Both sample data sets of Breast Cancer were reported to be sequenced using Illumine sequencer and were paired-end type. In the present investigation, the hardware and platform used for WES pipeline include a minimum 8GB RAM with a working platform Ubuntu operating system.
Quality analysis of raw data
The quality analysis of downloaded raw data samples was performed using the FASTQC tool.19 FastQC generates an HTML formatted report with box plots and graph plots for mean quality scores for sequences, GC content distribution, read length distribution, sequence duplication level and detects the overrepresented sequences indicating the contamination of adapter or primer.
Data Preprocessing
The preprocessing was performed for low-quality bases that are expected to be present at the ends of the reads which includes removal of adapter sequences and trimming of these low-quality bases.
Alignment to reference genome
The next step is the alignment of datasets with the reference genome hg38 Homo sapiens downloaded from the UCSC genome web browser. The reference genome of Homo sapiens is 3.5 GB file, for this index is built using the Bowtie2 index option that generates 6 files which are used for the alignment of samples with the reference genome.
Post-processing alignment
The SAM format was converted to BAM (Binary Alignment Mapping) format using SAM tools call the program.20 The SAM tools sort program was used to generate the sorted BAM file followed by generating indices. Then to remove the duplicates in sequence reads Picard tool’s20Mark Duplicate program is used.
Variant calling
Variant calling involves identifying the genomic variants which were done in two steps using SAM tools mpileup program and BCF tools.21 SAM tool’s mpileup program computes the likelihood of aligned reads of the samples and stores the likelihoods in mpileup format. The BCF tools call command uses the genotype likelihoods generated from the previous step to call SNPs and indels, and output all identified variants in the Variant Call Format (VCF).
Annotation
To call the variants in data samples, SIFT (Sorting Intolerant from Tolerant) 4G annotator22was used where SIFT predicts an amino acid substitution affecting protein function. The resulting xls format file was used to find the biological process affecting from the gene also the location of gene.
RESULTS
Quality Analysis of Samples
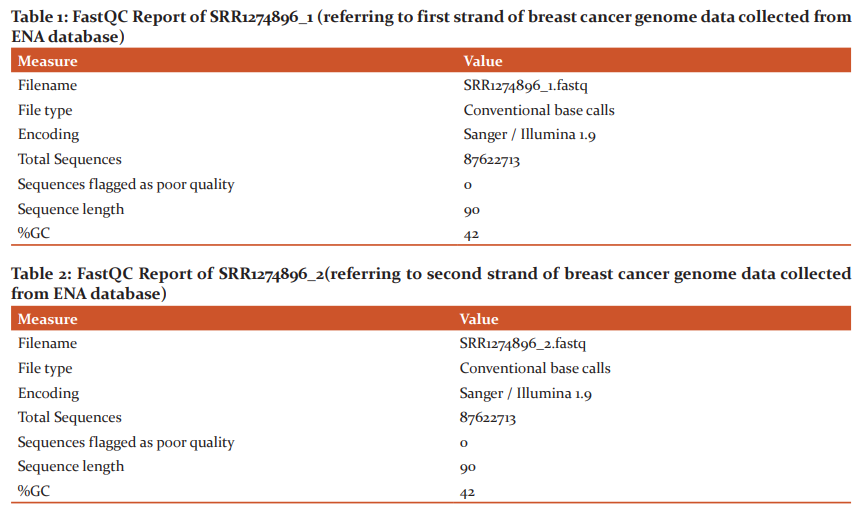
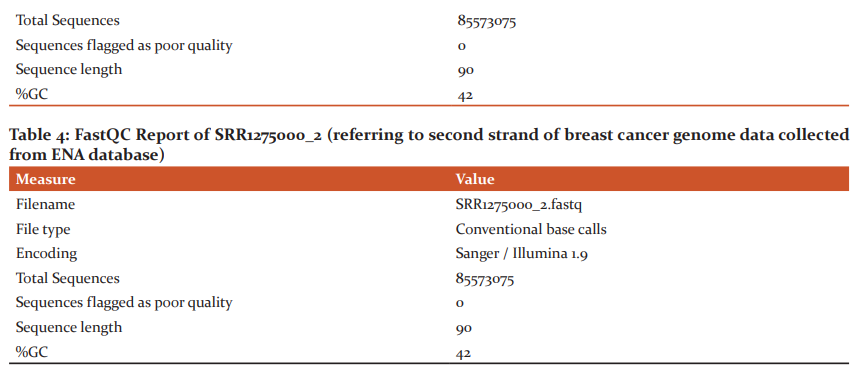
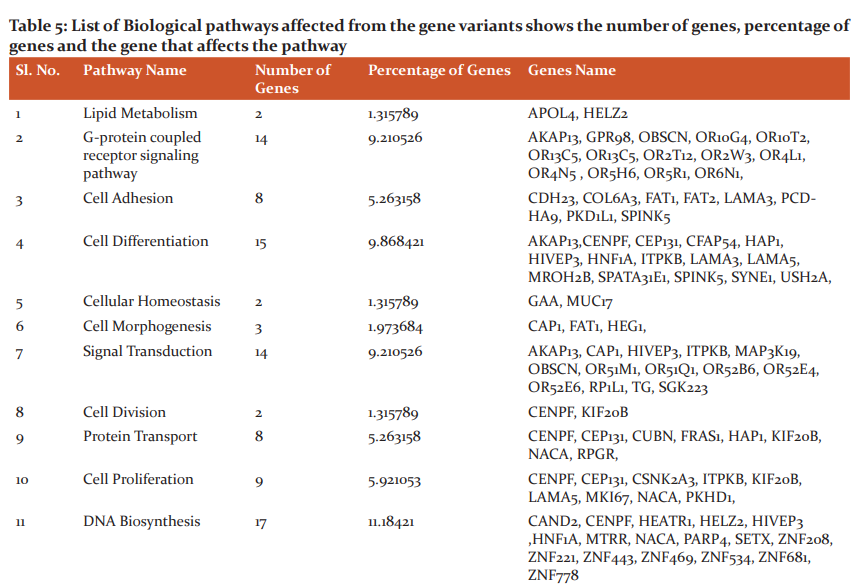
The raw samples downloaded from the ENA database were subjected to quality check using the FastQC tool, which generates HTML formatted report with box plots and graph plots for mean quality scores for sequences, read length and depth along with the intended coverage and overrepresented sequences. These results of quality analysis are shown in Table1 –Table4. Since there are no overrepresented sequences in the samples of breast cancer, further removal and trimming of the low-quality bases step was not performed for any samples.
Alignment Summary
The reads were aligned to the UCSC reference human genome hg38 using the sequence alignment tool Bowtie2. Both the samples of paired-end datasets are used for the alignment. The overall summary of alignment is listed below:
SRR1274896_1.fastq -2 and SRR1274896_2.fastq
87622713 reads; of these:
87622713 (100.00%) were paired; of these:
856366 (0.98%) aligned concordantly 0 times
70510403 (80.47%) aligned concordantly exactly 1 time
16255944 (18.55%) aligned concordantly >1 times
----
856366 pairs aligned concordantly 0 times; of these:
150735 (17.60%) aligned discordantly 1 time
----
705631 pairs aligned 0 times concordantly or discordantly; of these:
1411262 mates make up the pairs; of these:
849837 (60.22%) aligned 0 times
254711 (18.05%) aligned exactly 1 time
306714 (21.73%) aligned >1 times
99.52% overall alignment rate
The alignment summary consists of three sections;
-
Concordant alignment: In the above alignment result the (70510403 + 16255944) reads align concordantly which is 99.02% of reads.
-
Discordant alignment: The remaining 856366 reads is 0.98% (100-99.02%), of these, 150735 reads align discordantly. Then, of the non-concordant fraction, 17.60% of reads (150735 reads) align discordantly.
-
The remaining alignment whether concordant or discordant but both are aligned in paired-end mode. The rest of the reads either align as singles i.e. Read1 in one locus & Read2 in a completely different locus or one mate aligned and the other unaligned or may not align at all. Hence the reads that are in last section is Total-(concordant+ discordant).This is shown as 87622713-(86766347+150735) = 705631 reads. This alignment is in single fashion so calculate inmates (readsx2).
-
To reach the overall alignment, count the mates in total (i.e. mates aligned in paired and mates aligned in single fashion). That would be (86766347 X 2+ 150735X2) + 254711 + 306714= 174395589mates. That is 174395589 mates aligned of total (87622713 X 2) =175245426 mates, which is 99.52%.
SRR1275000_1.fastq and SRR1275000_2.fastq
85573095 reads; of these:
85573095 (100.00%) were unpaired; of these:
527907 (0.62%) aligned 0 times
65152529 (76.14%) aligned exactly 1 time
19892659 (23.25%) aligned >1 times
99.38% overall alignment rate
The above alignment was obtained in unpaired format which flagged 85573095 reads. In the above alignment, (65152529 + 19892659) = 85045188 aligned reads with coverage of 99.39%. Considering the overall alignment including 85045188 reads versus 85573095 showed 99.38% coverage.
Variant analysis
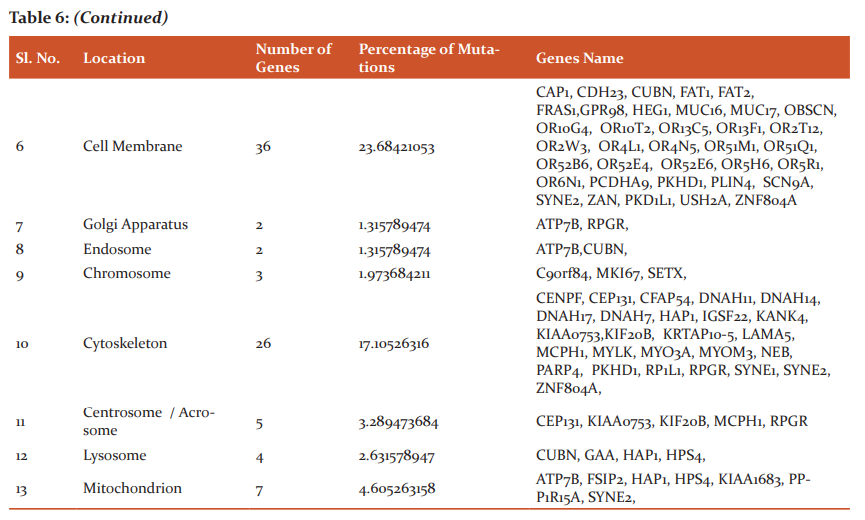
The file from the alignment step will be in SAM format in turn stored into BAM format this is followed by the generation of sorted file and removal of duplicates is done. In variant analysis .xls file is generated from the SIFT Annotator which is used for further work. The generated .xls consists of columns chromosome name, position, reference allele, alternate allele, Transcript ID, Gene ID, Gene Name, Region, Variant type, Reference Amino Acid, alternate Amino Acid, Amino acid position, SIFT score, SIFT median, number of sequences, dbSNP and SIFT prediction. The variant types of genes involved in causing cancer are Non-synonymous, Non-coding, Frameshift Deletion, Frameshift Insertion, Synonymous, Substitution, Non-Frameshift Deletion, Non-Frameshift Insertion, Start Lost, Stop Loss And Stop Gain. Thesample1 (SRR1274896_1.fastq and SRR1274896_2) consists of 40,598 and sample2 (SRR1275000_1.fastq and SRR1275000_2.fastq) consists 33,307 novel genes that causes breast cancer that are resulted in the dbSNP column. There were9,985 and 9,325 non-synonymous genes observed in sampl1 and sample2 respectively that are involved in causing Breast cancer. The results of variant annotation with the non-synonymous variant type of sample1 and sample2 were tabulated; among these mutations, variant type with 5 and above were listed out. The common genes from both samples possessing 5 mutations or more were chosen and the interpretation study of all common genes is carried out using Uniprot database that describes the pathway analysis, domain analysis, gene involvement in biological process and gene function. The interpretation study of genes showed biological pathways affected from the gene variants tabulated in Table5. Further investigation is made to know the tissue-specific location of genes which is depicted in Table 6.
DISCUSSION
We made an attempt to analyze the gene mutation found to occur in exome of Breast cancer. Pan et al. suggested that about 50-90% of risk of breast cancer are due to the mutations with BRCA1 or BRCA2.23 Timoteo et al. reported that rare male breast cancer risk was related to the BRAC2 gene deletion was shown using Exome sequencing.24 Thompson et al. reported that exome sequencing identified the DNA repair FANCC and BLM genes which are predisposing to breast cancer.25 Johanna et al. reported FANCM gene which is susceptible for triple-negative breast cancer using WES.26 Noh et al. using exome sequencing demonstrated 7 genes with mutations XCR1, DLL1, TH, ACCS, SPPL3, CCNF and SRL may be associated in causing breast cancer.27 Sarah et al. discovered gene SF3B1 mutation as a new target for anticancer therapy using exon sequencing and the whole genome sequencing methods.28
The result of present investigation revealed that the existence of 10314 synonymous mutation in the sample1 and 9826 in sample2indicating that such mutations are coding regions that does not change the protein sequence and amino acid does not change, then the protein also remain unaffected. The existence of 9985 non-synonymous mutations in sample1 and 9325 in the sample2 indicates such mutations occur due to insertion or deletion of single nucleotide in sequence or mutation changes the single nucleotide into a codon that does not translate into the same amino acid. The existence of 6619 Frameshift deletion in sample1 and 2712 in sample2; also 7631 Frameshift insertion in sample1 and 3324 in sample2 indicates such mutations arise when the normal sequence of codons is disrupted by the insertion or deletion of one or more nucleotides, provided that the number of nucleotides added or removed is not a multiple of three and cause the reading frame to shift. The existence of 157 non-frameshift insertions in sample1 and 42 in sample2; also130 non-frameshift deletion in sample1 and 56 in sample2 indicate the mutations arises due to insertion or deletions of 3 or multiples of 3 nucleotides that do not cause frameshift changes in protein-coding sequence. The existence2890 mutation in the sample1 and 1257 number substitution mutation in sample2indicates mutations arise by substituting a single nucleotide with different nucleotide. The existence of 27 and 23 number start lost mutation in sample1 and sample2 respectively indicate, that mutations occurs when start codon prevents the original start translation site from being used. The existence of 42 stop loss mutations in sample1 and 41 in the sample2 indicates stop-loss mutation is the loss of normal stop codon by the mutation. The existence of 107 stop gain mutations in sample1 and 115 in the sample2 indicate stop gain mutation is due to the creation of stop codon. The existence of 222806 non-coding mutation in sample1 and 153407 noncoding mutations in sample 2 indicating noncoding mutation indicating nucleotide do not encode protein sequences.
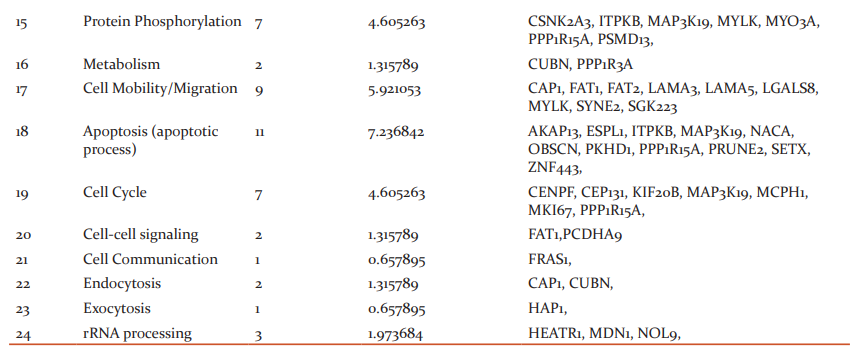
There were 211 and 179 genes that were non-synonymous, and showed 5 and above mutations in sample1 and sample respectively. The interpretation study was carried out on the 159 common genes among these mutations. Important genes considered through this study, AKAP13 reported to involve in G-protein coupled receptor signaling pathway, Cell Differentiation, Signal Transduction, Apoptosis playing an important role in assembling signaling complexes downstream of many different types of G protein-coupled receptors, APOL4 involved in Lipid Metabolism playing an important role in lipid exchange and transport throughout the body, GPR98, OR10G4, OR10T2, OR13C5, OR13C5, OR2T12, OR2W3, OR4L1, OR4N5, OR5H6, OR5R1 and OR6N1are reported to involve in G-protein coupled receptor signaling pathway, OBSCN is reported to involve in G-protein coupled receptor signaling pathway, Signal Transduction, Protein Biosynthesis and Apoptosis playing role in myofibrillogenesis. CDH23, COL6A3 and PKD1L1 involved in Cell Adhesion, FAT1 involved in Cell Morphogenesis, Cell Adhesion, Cell Mobility/Migration and Cell-cell signaling playing role for cellular polarization, directed cell migration and modulating cell-cell contact, FAT2 is reported to involve in Cell Adhesion, Cell Mobility/Migration playing role in the regulation of cell migration. LAMA3 involve in Cell Adhesion, Cell Differentiation, and Cell Mobility/Migration and role in mediating the attachment, migration and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components,PCDHA9 involve in Cell Adhesion, Cell-cell signaling helps in the establishment and maintenance of specific neuronal connections in the brain, SPINK5 involve in Cell Adhesion, Cell Differentiation, and Immune Responseplays role in the immune system by producing white blood cells called lymphocytes, CENPF is reported to involve in Cell Differentiation, Cell Division and Protein Transport, Cell Proliferation, DNA Biosynthesis, Protein Biosynthesis, Cell Cycle helps in chromosome segregation during mitosis, CEP131 involved in Cell Differentiation, Protein Transport, Cell Proliferation, Protein Biosynthesis and Cell Cycle helps in sperm flagella formation,CFAP54, MROH2B, SPATA31E1 and SYNE1 are reported to involve in Cell Differentiation, HAP1 is reported to involve in Cell Differentiation, Protein Transport, Protein Biosynthesis and Exocytosis playing role in intracellular trafficking or organelle transport. ITPKB is reported to involve in Cell Differentiation, Signal Transduction, Cell Proliferation, Protein Phosphorylation and Apoptosis helps in regulating the levels of a large number of inositol polyphosphates that are important in cellular signaling, LAMA5 involved in Cell Differentiation, Cell Proliferation, Protein Biosynthesis, and Cell Mobility/Migration playing a role in mediating the attachment, migration and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components. GAA, MUC17 are involved in Cellular Homeostasis, CAP1 is reported to involve in Cell Morphogenesis, Signal Transduction, Endocytosis, and Cell Mobility/Migration plays part in regulating filament dynamics and has been concerned in many complex developmental and morphological processes that include mRNA localization and establishment of cell polarity.HEG1 is involved in Cell Morphogenesis. HIVEP3 is reported to involve in Cell Differentiation and Signal Transductionhelpsas a transcription factor and binds the kappaB motif in target gene and regulates nuclear factor kappaB-mediated transcription, MAP3K19 is reported to involve in Signal Transduction, Protein Phosphorylation, Apoptosis and Cell Cycle play role in regulating TGF-β-induced Smad signaling and gene expression.OR51M1, OR51Q1, OR52B6, OR52E4, OR52E6, RP1L1 and TG are reported to involve in Signal TransductionSGK223 is involved in Signal Transduction and Cell Mobility/Migration. KIF20B is reported to involve in Cell Division, Protein Transport, Cell Proliferation, Protein Biosynthesis and Cell Cycle playing role in regulating neuronal polarization.CUBN is reported to involve in Protein Transport and Endocytosis where role is the metabolism of vitamins, lipoprotein and iron by facilitating their uptake. FRAS1 and RPGR are involved in Protein Transport and Protein Biosynthesis, NACA is reported to involve in Protein Transport, Cell Proliferation, DNA Biosynthesis, Apoptosis and Protein Biosynthesis plays its role in ventricular cardiomyocyte expansion and regulates postnatal skeletal muscle growth and regeneration. CSNK2A3 is reported to involve in Cell Proliferation, MKI67 is reported to involve in Cell Proliferation and Cell Cycle helps in chromatin organization. CAND2 is involved in DNA Biosynthesis and Protein Biosynthesis playing role in the cellular repertoire of SCF (SKP1-CUL1-F-box protein) complexes, HELZ2 involved in DNA Biosynthesis and Lipid Metabolism helps in transcriptional coactivator for nuclear receptors. PPARA, PPARG, THRA, THRB and RXRA. MTRR, PARP4, ZNF208, ZNF221, ZNF534, ZNF681 and ZNF778 are reported to involve in DNA Biosynthesis, ZNF443 is reported to involve in DNA Biosynthesis and Apoptosis CHGB, HPS4, HPS4, KIAA0753, METTL22 and TTN are reported to involve in Protein Biosynthesis.PSMD13 is involved in Post Translation Modification and Protein Phosphorylation helps in maintaining protein homeostasis by removing misfolded or damaged proteins that may impair cellular functions. CRACR2A, GBP6 are reported to involve in Immune Response, CSNK2A3, MYO3A are reported to involve in Protein Phosphorylation, MYLK is involved in Protein Phosphorylation and Cell Mobility/Migration helps in regulation of epithelial cell survival. PPP1R15A is involved in Protein Phosphorylation, Apoptosis, LGALS8, SYNE2, ESPL1 and PRUNE2 are reported to involve in Cell Mobility/Migration SETX is involved in DNA Biosynthesis and Apoptosis plays role in transcription regulation by modulating RNA Polymerase II (Pol II) binding to chromatin and through interaction with proteins involved in transcription, FRAS1 is involved in Cell Communication helps in anchoring the top layer of skin by connecting the basement membrane of the top layer to the layer of skin below. MDN1, NOL9 are involved in rRNA processing.
Mutations
There are 5566 and 3611 genes showed less than 5 mutations in sample1 and sample2 respectively and were considered to be non-potential gene variants. 112 Genes showed 5mutations, 48 genes showed 6 mutations, 27 genes showed 7 mutations, 14 genes showed 8 mutations, 10 genes showed 9 mutations,10 genes showed 10 mutations, 3 genes showed 11 mutations,12 mutations and 13 mutations, 2 genes showed 14 mutations,15 mutations, 19 mutations, MKI67 showed 17 mutations, CENPF showed 20 mutations, MUC17showed 23 mutations, AHNAK2 showed 32 mutations, TTN showed 34 mutations, FLG showed 36 mutations, MUC16 showed 46 mutations, MUC3A showed 55 mutations in sample1. In the sample2, 88 Genes showed 5 mutations, 35 genes showed 6 mutations, 22 genes showed 7 mutations, 7 genes showed 8 mutations, 10 genes showed 9 mutations, 6 genes showed 10 mutations, 5 genes showed 11 mutations, 2 genes showed 20 mutations and 30 mutations, XIRP2 showed12 mutations AL
PK2 showed 13 mutations, GPRIN2 showed 15 mutations, MKI67 showed 17 mutations, OBSCNshowed 19 mutations, TTN showed 34 mutations, MUC16 showed 45 mutations, MUC3A showed 50.
The gene mutations with 5 and above were considered to be potential gene variants used for further analysis. The MUC3A showed the highest mutations in both samples making it potential gene variant observed in breast cancer conditions. Williams et.al, reported that expression of MUC3A is down-regulated in colorectal cancer.29 Rakha et al. concluded that theMUC3A expresses in human breast cancer tissues.30 Leroy et al. Park et al. Wang et al. Rakha et al. concluded that MUC3A is poor prognosis, that has been shown in pancreatic, breast, gastric, and renal cancers.30-33 The MUC16 showed 45 mutations in both samples. Das et al. stated that MUC16 plays role in tumour genesis and metastasis for ovarian cancer, pancreatic cancer and breast cancer.34Mildred et al. reported that MUC16 has C125 biomarker plays role in ovarian tumour growth and metastasis.35
Biological Pathways Affected
An interpretation study was carried on common genes and study revealed the genes that affect the biological pathways and location of the mutated genes. 17 genes affected DNA biosynthesis and protein biosynthesis, 15 genes were found to be affecting cell differentiation, 14 genes were found to be affecting G-protein coupled receptor signaling pathway and signal transduction, 11 genes affected Apoptosis, 9 genes affected cell mobility/migration and cell proliferation, 8 genes affected Cell adhesion and protein transport, 7 genes were found to be affecting protein phosphorylation and cell cycle, 3 genes were found to be affecting genes cell morphogenesis, immune response and rRNA processing, 2 were found to be affecting genes were found to be affecting lipid metabolism, cellular homeostasis, cell division, post-translation modification, metabolism, cell-cell signaling and endocytosis, single gene were found to be affecting cell communication and exocytosis.
In the present study, we observed the location of the 42 mutated genes product in Cytoplasm, 39 in nucleus, 36 mutated gene products were in cell membrane, 27 were in the extracellular region, 26 gene products in extracellular matrix and cytoskeleton, 7 gene products found in the mitochondrion,5 gene products found in centrosome/acrosome, 4 inlysosome, 3 in the chromosome, 2 in golgi apparatus and endosome, 1 in cell cortex.
CONCLUSION
Breast cancer occurs when a malignant tumor originates in the breast. As breast cancer tumors mature, they may spread to other parts of the body. In the current work WES analysis of gene variants involved in causing breast cancer were studied. The results revealed that 5,580,674 genes were found in sample1 and 3,850,125 found in sample2. Among these genes, 9985 and 9325 genes were non-synonymous in samples 1 and sample2 respectively. 5566 genes possessed less than 5 mutations in the sample1 and 3611 showed in sample2. These genes were considered to be non-potential gene variants, hence were not used for study. 211 genes were non-synonymous with 5 and above mutations in the sample1 and 179 in sample2. Among these two samples 153 genes were common and an interpretation study was made on these common genes. The study revealed that MUC3A possessed a maximum number of mutations in both the samples (55 in sample1 and 50 in sample2) indicating potential gene variants in breast cancer condition. This MUC3A can be used as a biomarker for finding treatment for breast cancer. Also one more mucin, MUC16 showed 46 mutations in both samples. The biological process study revealed that 17 genes affect DNA and protein biosynthesis. Also the location of these genes showed those42 mutated genes in the cytoplasm and 39 mutated genes in the nucleus indicating the impact of gene variation on the intracellular process.
ACKNOWLEDGEMENTS
The authors express their sincere gratitude to all the authors/ editors/publishers of those articles, journals, and books from where the literature of this article has been reviewed and discussed. Also authors especially thank Dr Sandhya Kulkarni, Principal KLE’s GH College for her invaluable support. All the authors want to thank all those who participated in this study.
Conflict of Interest: All authors confirm that there are no conflicts of interest regarding this article.
Source of funding: This work does not receive any funding.
Author’s Contribution:
-
Dr. Dhanyakumar G : Clinical aspect of Brest Cancer
-
Dr. Maheswari L Patil: Collection of Samples from databases, Whole Exome analysis of Breast cancer samples, article write-up.

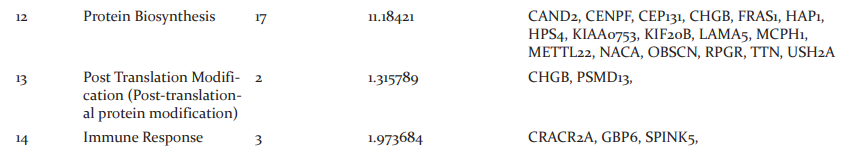

References:
-
Parkin DM. International variation. Oncogene 2004; 23: 6329-6340.
-
De Jong MM., Nolte IM., Merman GJ, van derGraaf WT, Oosterwijk JC, Kleibeuker J H, et al. Genes other than BRCA1 and BRCA2 involved in breast cancer susceptibility. J Med Genet 2002; 39:225-242.
-
Gorski B, Debniak T, Masojc B, Mierzejewski M, Medrek K, Cybulski C, et al. Germline 657del5 mutation in the NBS1 gene in breast cancer patients.Int J Cancer2003; 106:379-381
-
Ergul E., Sazci A., Utkan Z., Canturk N. Z. Polymorphisms in the MTHFR gene are associated with breast cancer. Tumour Biol 2003; 24: 286-290.
-
Goode E. L., Ulrich C. M., Potter J. D. Polymorphisms in DNA Repair Genes and Associations with Cancer Risk. Cancer Epidemiology, Biomarkers Prev 2002; 11: 1513-1530.
-
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011; 13(6):224.
-
Dietze EC, Sistrunk C, Miranda-Carboni G, O’Regan R, Seewaldt VL. Triple-negative breast cancer in African-American women: disparities versus biology. Nat Rev Cancer 2015; 15(4):248-254.
-
Wang Y, Zhai X, Liu C, Wang N, Wang Y. Trends of triple-negative breast cancer research (2007–2015): A bibliometric study. Medicine (Baltimore) 2016; 95(46):e5427.
-
Jiao Q, Wu A, Shao G., Peng H., Wang M, Ji S et al. The latest progress in research on triple-negative breast cancer (TNBC): risk factors, possible therapeutic targets and prognostic markers. J Thorac Dis. 2014; 6(9):1329-1335.
-
Mehrgou A., Akouchekian M. The importance of BRCA1 and BRCA2 genes mutations in breast cancer development. Med J Islam Repub Iran 2016 15; 30:369.
-
Petrucelli N, Daly MB, Feldman GL. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Gene Med. 2010; 12(5).
-
Ungerleider NA, Rao SG., Shahbandi A, Yee D, Niu T, Frey WD et al. Breast cancer survival predicted by TP53 mutation status differs markedly depending on the treatment. Breast Cancer Res. 2018; 115.
-
Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. HumMol Genet.1998;(7):507–15.
-
Tavusbay C, Acar T, Kar H, Atahan K, Kamer E. Patients with Peutz-Jeghers syndrome have a high risk of developing cancer. Turk J Surg 2018; 34(2):162-164.
-
Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature1998;(391):184–7.
-
Apostolou P, Fostira F. Hereditary Breast Cancer: The Era of New Susceptibility Genes. BioMed Res Int. 2013.
-
Kuang Y, Siddiqui B, Hu J, Pun M, Cornwell M, Buchwalter G, et al. Unraveling the clinicopathological features driving the emergence of ESR1 mutations in metastatic breast cancer. Breast Cancer 2018; 4:22
-
Sun YS, Zhao Z, Yang ZN, Xu F, Lu HJ, Zhu ZY, et al. Risk Factors and Preventions of Breast Cancer. Int J Biol Sci. 2017; 13(11):1387-1397.
-
Andrews S. FastQC. Babraham Bioinformatics; 2012. Cambridge, UK.
-
Bao R, Huang L, Andrade J, Tan W, Kibbe WA, Jiang H, et al. Review of Current Methods, Applications, and Data Management for the Bioinformatics Analysis of Whole Exome Sequencing. Cancer Infor. 2014; 13(Suppl 2):67–82.
-
Bonfield JK, Marshall J, Danecek P, Li H, Ohan V, Whitwham A et al. HTSlib: C library for reading/writing high-throughput sequencing data. GigaScience,2021; 10(2)
-
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protocols 2106; 11: 1-9.
-
Pan H, He Z, Ling L, Ding Q, Chen L, Zha X, et al. Reproductive factors and breast cancer risk among BRCA1 or BRCA2 mutation carriers: results from ten studies. Cancer Epidemiol. 2014; 38(1):1e8.
-
Timoteo ARdS, Albuquerque B.M, Moura PPP, Ramos CCdO, Agnez-Lima LF, et al. Identification of a new BRCA2 large genomic deletion associated with high-risk male breast cancer. Here Cancer Clin Pract 2015; 13(1):2.
-
Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genetics 2012; 8(9).
-
Kiiski JI, Pelttari L.M., Khan S, Freysteinsdottir ES, Reynisdottir I, Hart SN, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc Natl Acad Sci USA 2014; 111(42):15172e15177.
-
Noh JM, Kim J, Cho DY, Choi DH, Park W, Huh SJ. Exome sequencing in a breast cancer family without BRCA mutation. Radiat Oncol J. 2015; 33(2):149e154.
-
Maguire SL, Leonidou A, Wai P, Marchiò C, Ng CK, Sapino A, et al.SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol 2015; 235(4):571e580.
-
Williams SJ, McGuckin MA, Gotley DC, Eyre HJ, Sutherland GR, Antalis TM,Two novel mucin genes down-regulated in colorectal cancer identified by differential display.Cancer Res 1999; 59(16):4083-9.
-
Rakha EA, Boyce RW, Abd El-Rehim D., Kurien T., Green AR., Paish EC., et al. Expression of mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and their prognostic significance in human breast cancer. Mod Pathology2005; 18(10):1295-304.
-
Leroy X, Gouyer V, Ballereau C, Zerimech F, Huet G, Copin MC, et al. Quantitative RT-PCR assay for MUC3 and VEGF mRNA in renal clear cell carcinoma: relationship with nuclear grade and prognosis.Urology2003; 62(4):771-5.
-
Park HU., Kim J.W., Kim G.E., Bae H.I, Crawley SC, Yang SC, et al. Aberrant expression of MUC3 and MUC4 membrane-associated mucins and sialyl Le(x) antigen in pancreatic intraepithelial neoplasia.Pancreas2003; 26(3):e48-54.
-
Wang RQ, Fang DC. Alterations of MUC1 and MUC3 expression in gastric carcinoma: relevance to patient clinicopathological features. J Clin Pathol 2003; 56(5):378-84.
-
Das S, Batra SK. Understanding the Unique Attributes of MUC16 (CA125): Potential Implications in Targeted Therapy. Cancer Res 2015; 75(22):4669-4674.
-
Felder M, Kapurl A, Gonzalez-Bosquet J, Horibatal S, Heintz J, Albrecht R, et al. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer 2014, 13:129.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





