IJCRR - 14(3), February, 2022
Pages: 31-35
Date of Publication: 01-Feb-2022
Print Article
Download XML Download PDF
Problematic Patterns of HPLC in Common Practice: Decision Making and Further Investigations
Author: Hota Sarbashis, Das Tushar Kanti
Category: Healthcare
Abstract:Introduction: HPLC (High Performance Liquid Chromatography) is the commonest method endorsed for screening of Thalassemia and other hemoglobinopathies in our country. Universal screening of antenatal mothers in the early first trimester has been employed under the Thalassemia control programme launched as a part of National Health Mission. Estimated prevalence of thalassemia in India is as high as 10000-15000 per year and varies from state to state, and even district to district owing to the multi-ethnic population of India and the scattered tribal belts. Case Reports: Here, our discussion has highlighted four scenarios which are reasonably common in Indian subcontinent, but problematic none-the-less due to various reasons. The differential diagnoses, decision making and strategies for further choice of investigations has been elaborated in details. Conclusion: As a tool of screening, the outcome based on haemoglobin HPLC greatly relies on the successful interpretation of the patterns of the graphs and thorough understanding of the gray zones and pit falls inherent to the procedure employed.
Keywords: : Borderline HbA2 value, E-beta thalassemia, Sickle-beta thalassemia, alpha thalassemia carrier, HPLC, Beta-thalassem�ia carrier
Full Text:
Introduction:
HPLC (High Performance Liquid Chromatography) is the commonest method endorsed for screening of Thalassemia and other hemoglobinopathies in our country. It has the added advantage of quantification of the variant hemoglobin in the same sitting, which was not achieved by previous electrophoresis based methods.1 Universal screening of antenatal mothers in the early first trimester has been employed under the Thalassemia control programme launched as a part of National Health Mission.2 Estimated prevalence of thalassemia major in India is as high as 10000-15000 per year and varies from state to state2, and even district to district owing to the multi-ethnic population of India and the scattered tribal belts.
As a tool of screening, the outcome based on haemoglobin HPLC greatly relies on the successful interpretation of the patterns of the graphs and thorough understanding of the gray zones and pit falls inherent to the procedure employed. Here, our discussion will highlight those scenarios which are reasonably common in Indian subcontinent, but problematic none-the-less due to various reasons. The differential diagnoses, decision making and strategies for further choice of investigations will be elaborated in details.
Case series:
1) Age- 18 yr, Hb- 10.3 gm%, RBC count- 3.83 million/mm3, HCT-31.50%, MCV- 82.20 fl, MCH-26.90 pg, RDW-18.50 %
Impression: The person was asked to repeat HPLC after Iron-folate supplementation for 3 months, as the iron profile revealed a low Ferritin level and the result of Vit B12 & Folate assay were below the reference range. Subsequent HPLC confirmed a Beta-Thalassemia carrier state.
2) Age- 36 yr, Hb- 4.20 gm%, RBC count- 2.13 million/mm3,HCT-15.70%, MCV- 73.70 fl, MCH-19.70 pg, RDW-42.40 %
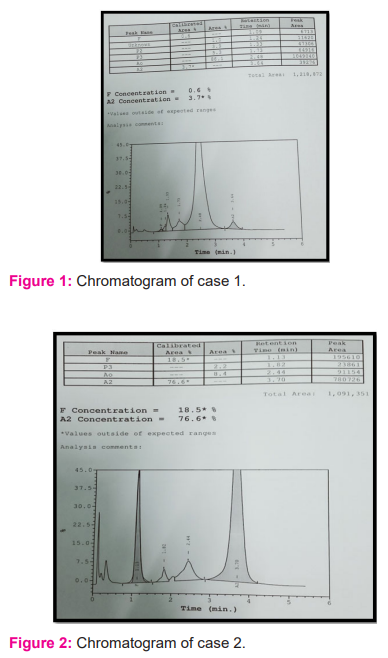
Impression: compound heterozygote of Hb E and Beta-thalassemia.
3) Age- 8 yr, Hb- 9.2 gm%, RBC count- 3.87 million/mm3,HCT-28.9%, MCV- 74.7 fl, MCH-23.8pg, RDW-21.8
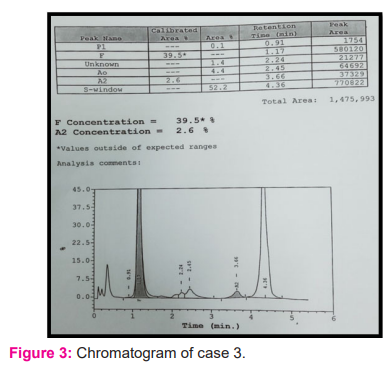
Impression: Compound heterozygote of Beta-Thalassemia and Sickle Hemoglobin (Hb S). A genetic study was advised for confirmation as well as to rule out a coexistent alpha thalassemia trait.
4) Age- 19 yr, Hb- 11.9 gm%, RBC count- 4.22 million/mm3,HCT-34.90%, MCV- 82.7fl, MCH-28.1pg, RDW-13.60 %
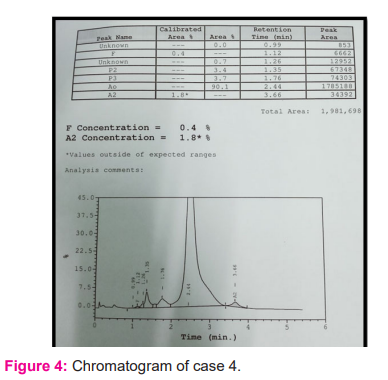
Impression: Patient was referred for genetic study to rule out an alpha-thalassemia trait.
Discussion:
The commonly used software is the Beta Thalassemia short programme designed by Bio-Rad, which mainly focuses on screening of beta-thalassemia syndromes; and thus the first 63 seconds are not integrated in the graph.3 So the abnormal hemoglobin important in alpha thalassemia detection (like HbH and Hb Bart) cannot be visualised in it. The pre-analytical quality control is also of paramount importance and the EDTA blood samples freshly collected should be run as early as possible; as storage-related deterioration can manifest as multiple aberrant peaks(commonly at P3), which are very difficult to interpret. Another important matter is the measurement of total calibrated area, which ideally should remain between 1 million to 3 million for a properly validated result.
It is to be remembered that what we actually detect in HPLC- is the altered pattern of Hb A and HbF percentage in case of adults, which indirectly points toward the diagnosis of thalassemia syndrome. The HPLC-based diagnosis is mostly impractical in infants, where the shift from fetal to adult hemoglobin has not been completed yet4. Moreover, the hemoglobin shift is often delayed in cases of children with hemoglobinopathies, and that creates further confusion.
Children with clinically diagnosed beta-thalassemia major often receive multiple transfusions for obvious reasons, prior to a definitive diagnosis is made. The HPLC pattern generated from such patients sustained on repeated transfusions create awesome problem regarding interpretation4, and often the molecular diagnosis is the only resort available.
-
Borderline value of HbA2 in a suspected beta-thalassemia trait: A common problem is encountered most often when the HbA2 percentage falls between 3.5% and 4% just like in case 1.
It is important to remember there are entities like Silent and almost Silent beta-thalassemia carrier, where the HbA2 percentage remains normal or not much elevated.5It is impossible to diagnose a carrier state in such scenarios without any genetic study. Co-inheritance of delta thalassemia or alpha thalassemia decreases the level of Hb A2 that was likely to be increased due to a carrier state5.
Perhaps the most important confounding factor in Indian perspective, especially for antenatal screening, is Iron deficiency and megaloblastic anaemia. It is a dictum to scrutinize the hemoglobin, RBC count and red cell indices in details and assess the iron profile, vit B12 & folate levels of the patient, in such cases. Repeat HPLC is to be done after iron-folate supplementation, only then the carrier state may become manifest.
The presence of Hb A2 variants, often which elutes in the S window, may underestimate the actual value of A2, and, thus a true thalassemia carrier can be missed5.
In our case, the red cell indices with the borderline HbA2 value raised suspicion for a beta-thalassemia carrier state. An iron-profile and vitamin B12 and folate assay were sought. The person was asked to repeat HPLC after Iron-folate supplementation for 3 months, as the iron profile revealed a low Ferritin level and the result of Vit B12 & Folate assay were below the reference range. Subsequent HPLC confirmed a Beta-Thalassemia carrier state.
-
E-beta thalassemia or Hb E disease: The common problem in HPLC Bio-Rad II is the co-elution of different hemoglobins in the same window. It is particularly important for the A2 window, where Hb Lepore and Hb E also appear. Hb E is very common in the Indian subcontinent, especially in the Eastern India surrounding Bengal2. No other common test can separate Hb A2 and Hb E, except Capillary Electrophoresis; although it has the disadvantage of separating Post-translationally modified variants of the same haemoglobin, and thus makes the interpretation troublesome.1 It is also important to study the graph in these cases, otherwise, the Lepore hump can be missed.
It is taken as a thumb rule that whenever the HbA2 percentage crosses the value of 10%, some other Hemoglobin like Lepore or E are likely present.1 The problem lies in the fact that actually, no clear cut line of separation is existent, between the diagnosis of E-beta thalassemia and E-disease based on HbA2 percentage only. There is a region of overlap between 65% and 85% of HbA2 percentage, which is shared by both.
The common strategy here is to look for the Hb F percentage, usually, it is less than 5% in Hb E disease but more than 15% in cases of A-beta thalassemia. But still there remains a foggy area. In this scenario, the clinical data is of particular significance. Patients with E-beta thalassemia often have past history of multiple blood transfusions, but those with Hb E disease do not have such. The parental study, often, when feasible, ameliorate the problem, but when one of them is dead, the only resort left is genetic study.
In our case, the patient was symptomatic and had previous history of transfusions. The peripheral blood findings also favoured the diagnosis of E-beta thalassemia. The parents were alive in this case, and further HPLC-based study revealed the father being a beta-thalassemia carrier and the mother a Hb E trait.
-
Sickle cell disease versus Sickle Beta Thalassemia: Diagnosis of sickle cell disease is done when Hb S is greater than 50% and there is no adult hemoglobin (Hb A), as compared to Sickle cell trait when both Hb A and Hb S are present and the former is the predominant one. In Indian context, due to the presence of the Arab-Indian haplotype, Hb F comprises a significant proportion in cases of Sickle cell disease (maybe up to 40%).5
The diagnosis of compound heterozygosity of sickle hemoglobin and β+ thalassemia is usually straightforward.5 Both Hb A and Hb S are present, and the percentage of Hb S is greater than Hb A.
However, the problem arises regarding the distinction of Sickle-β0 thalassemia and Sickle cell anemia. In both cases, no adult hemoglobin is there and the HPLC results are almost similar. The HbA2 percentage can aid in diagnosis. Usually, in sickle- β0 thalassemia the HbA2 is found to be elevated (3.5-5.5%), whereas it remains at 2-4% in Sickle cell disease.5 The peripheral blood picture is of utmost importance in this case, microcytosis favors the diagnosis of Sickle-β0 thalassemia over Sickle cell disease. Also, the parental study, when feasible, can alleviate the problem.
However, if the case is Sickle cell anemia with microcytosis(with co-existent alpha-thalassemia), the differentiation is extremely difficult as the HbA2 percentages are almost similar and often need genetic diagnostic aids.5 A case of Sickle- β0 thalassemia has every chance to be misdiagnosed as sickle cell disease when coexistent alpha thalassemia is there.5
In our study, the HPLC pattern pointed towards the diagnosis of Sickle cell disease over Sickle-Beta thalassemia, as the HbA2 level was lower. However, the red cell indices created a strong suspicion for a coexistent beta-thalassemia. So a parental study was sought, luckily both were available. The mother was diagnosed as a case of sickle cell trait and the father, a beta-thalassemia carrier. The normal HbA2 level, in this case, was likely due to the co-existence of an alpha thalassemia trait; so, for confirmation, a genetic study was advised.
-
Can the case be that of an alpha thalassemia trait? It is very difficult to tell. Patients with Hb H disease (deletion of three alpha genes) present either in childhood with anemia, splenomegaly or the diagnosis is made incidentally in adult life. However, in both cases, the Hb H peak is found in HPLC and the diagnosis is straightforward. But, it should be borne in mind that the Hb H peak is not integrated in the beta-thalassemia short program and therefore likely to be missed if separately not searched for. The Hb%, peripheral smear findings and most importantly diminished A2 percentage in otherwise unremarkable HPLC should arouse prompt suspicion, to search for a suitable cause. Demonstration of Characteristic Golf ball inclusions by supra-vital stain can aid in diagnosis.1 It is also important to remember, that iron deficiency can completely mask the disease.5
α+ heterozygote(αα/α-) and α+ homozygote(α-/α-) cannot be detected by HPLC and most often Red cell indices are also normal.4 α0 trait (αα/--) is also very difficult to diagnose by HPLC but the altered red cell indices in the otherwise normal individuals should arise suspicion.
Molecular genetic study is the only resort for diagnosis here.4 As HPLC-based screening cannot efficiently diagnose the carrier state of alpha thalassemia, the occurrence of HbH disease and Hydropsfetalis cannot be prevented by HPLC-based screening protocol.
In our case, the red cell indices pointed towards a carrier state of thalassemia syndrome. Coupled with the diminished Hb A2 percentage, it gave rise to a strong suspicion as the iron-folate profile was almost normal, and the patient was referred for a genetic study to rule out an alpha-thalassemia trait.
Mutational Spectrum and molecular diagnosis:
Panigrahi I et al. have studied the mutational spectrum of thalassemia syndromes in India6, whereas Sinha et al. focussed on the regional and state-level diversity of the spectrum.71) IVS 1-5 G→C, 2) IVS 1-1 G→T, 3) mutation of codon 41/42, 4) mutation of codon 819, 5) 619 bp deletion are the common mutations found in case of beta-thalassemia. On the other hand, 1)-α4.2, 2)-α3.7, 3)-αSA, 4)Hb Koya Dora are the common aberrations found in alpha thalassemia. Mondal S K et al. have generated a valuable database on the prevalence of thalassemia in Eastern India, after studying more than a lakh cases over 10 years.8 The common method for molecular detection is usually based on PCR;- ARMS PCR for Beta-thalassemia and Gap PCR for alpha thalassemia.
The issue of sickle cell disease has been addressed by the study of Serjeant G R et al.9 Sen R et al. have drawn our attention to the prevalence of alpha thalassemia cases in the tribal belts of India.10
Conclusion:
It should remain clear what to expect from HPLC-based thalassemia screening and what to not. Although it is an efficient tool for screening beta-thalassemia traits, it cannot totally rule out the presence of a thalassemia carrier state, owing to the extreme heterogeneity of causative mutations and their more complicated genotype-phenotype correlation. Moreover, the alpha thalassemia traits are easy to miss. Ancillary studies including parental and sibling screening, other electrophoresis-based methods or genetic studies for mutation analysis are to be advocated in appropriate settings. The genetic studies may also reveal inconclusive results; as in most of the molecular biology labs, a handful of tools are employed to rule out only the most common genetic aberrations present, based on the local prevalence patterns.
Acknowledgment: Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to the authors/editors/publishers of all those articles, journals, and books from which the literature for this article has been reviewed and discussed.
Authors’ Contribution: Dr Sarbashis Hota- Conceptualization of the topic, Collection of Data, Review of Literature, manuscript writing.
Dr TusharKanti Das- Overall guidance and supervision, Design of the framework of manuscript, contributing author in the discussion, necessary correction and guidance for final submission.
References:
-
Bain B J, Bates I, Laffan M A. Dacie & Lewis Practical Haematology, twelfth edition. Elsevier, 2017
-
Prevention and control of Hemoglobinopathies in India- Thalassemias, Sickle cell Disease and another variant Hemoglobins-National Health Mission guidelines on hemoglobinopathies in India- Ministry of Health and Family Welfare, Govt of India, 2016.
-
Manual of Bio-Rad II HPLC machine, Bio-Rad India Private Limited, 2006.
-
Hemoglobinopathies: Current Practices for Screening, Confirmation and Follow up. Association of Public Health Laboratories. CDC. December 2015.
-
Bain B J, Haemoglobinopathy diagnosis, Third edition. Wiley Blackwell, 2020.
-
Panigrahi I, Marwaha R K. Mutational spectrum of thalassemias in India. Indian J Hum Genet. 2007 Jan-Apr; 13(1): 36-37.
-
Sinha S, Black M L, Agarwal S, Colah R, Das R, Ryan K et al. Profiling beta-thalassemia mutations in India at state and regional levels: implications for genetic education, screening and counseling programs. Hugo J. 2009 Dec; 3(1-4): 51-62.
-
Mondal S K, Mondal S. Prevalence of thalassemia and hemoglobinopathy in Eastern India: A 10-year high-performance liquid chromatography study of 119,336 cases. Asian J Transfusion Sci. Jan-Jun; 10(1): 105-110.
-
Serjeant G R, Ghosh K, Patel J. Sickle cell disease in India: A perspective. Indian J Med Res. 2016 Jan; 143(1): 21-24
-
Sen R, Chakrabarti S, Sengupta B, De M, Haldar A, Poddar S et al. Alpha –thalassemia among tribal populations of Eastern India. Hemoglobin. 2005; 29(4):277-80.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





