IJCRR - 14(2), January, 2022
Pages: 85-92
Date of Publication: 16-Jan-2022
Print Article
Download XML Download PDF
A Neuropharmacological Review of Alzheimer's Disease
Author: Shubham S. Bagade, Laxmikant B. Borse, Atul R. Bendale, Anil G. Jadhav
Category: Healthcare
Abstract:Alzheimer is one of the most frequent diseases that affect nerve cells in various sections of the brain. Pathologically, it occurs due to intracellular neurofibrillary tangles and extracellular amyloid protein depositions that result in the obstruction of neural transmission, culminating in this neurodegenerative illness. Additionally, food and nutrition are essential for developing and preventing Alzheimer's. The biomarker utilized for detecting the disease should be able to differentiate between different causes of dementia and detect it early. Use of Induced Pluripotent Stem Cells shows to be a successful treatment for the condition mentioned. There are three main hypotheses presented as a cause of AD: the cholinergic, tau and amyloid hypothesis. Additional risk factors include advancing age, genetics, head trauma, vascular illnesses, infections, and the environment in general. The two types of approved medications to treat AD (NMDA antagonists and cholinesterase inhibitors) are successful in treating the symptoms of AD, but are not cures or preventatives of the disease. Current AD research targets multiple processes, such as the aberrant tau protein metabolism, \?-amyloid, inflammatory response, and cholinergic and free radical damage, to find viable therapeutics capable of preventing or changing the progression of Alzheimer's disease. This review's purpose is to illustrate the pathway that leads to this condition and oncology treatment for it.
Keywords: Alzheimer’s disease, Acetyl cholinesterase inhibitors, N-methyl D-aspartate receptor antagonist, Beta amyloid, Neurofibrillary tangles
Full Text:
INTRODUCTION
Alzheimer’s Disease is the main cause of Dementia which contributes about 60% - 80% of total Dementia patients provided by WHO.1,10,13 Dementia is one of the prime causes in individuals around the globe for impairment and dependence. AD is a psychological condition that progresses with the age destroying memory, thinking skills, cognitive abilities and eventually the ability to carry out simple daily tasks. AD is an illness that impairs the central nervous system mainly affecting the temporal lobe, entorhinal cortex and hippocampus and in progression affecting the cerebral cortex of the brain which may be responsible for language, reasoning and social behavior leading to death with the advancement of the disease.1 The main condition in the disease is Amyloid plaques, neurofibrillary tangles and Lewy bodies appear in the brain.4 AD involves the medial temporal lobe, which houses the entorhinal cortex and hippocampus. Anterograde episodic memory loss is produced by the collapse of these mechanisms, and this shows up as forgotten daily minutiae.3,4
While the issue may seem benign, the symptoms may be seen by family members and the patient. Cognitive deficits which are severe enough to affect daily functioning are the current criteria for diagnosing AD (MCI- Mild Cognitive Impairment). An estimated 10% of MCI patients may develop AD annually.11,12 AD damages cognitive and functional ability over time, including visuospatial and executive function. The latter years of the disorder are associated with an increase dependency and neurological damage (akinetic mutism). Lack of mobility often results in 6–12-year deaths from lung or venous embolism. AD is diagnosed based on the patient's clinical findings. Neuroimaging is used to diagnose out those other maladies that could cause Alzheimer's-like symptoms. Shortly, laboratory tests, such as analysis of biomarkers, genetic testing, and molecular/functional neuroimaging, will likely be added into diagnostic criteria for AD to improve the sensitivity of diagnosis, notably in the initial and latter course of the disease.
Causes of AD are poorly analyzed as no perfect treatment is available to eradicate the disease. Only the progress of the disease can be slowed by medications and treatment. The pharmacological treatment available for AD is acetylcholinesterase inhibitors, antioxidants, NMDA channel blocker and other pharmacological treatments.6,9,14 The management of the disease and how medicines engage with the brain during treatment are crucial. Different aspects are discussed regarding the neuropharmacology of AD from the available literature.
HISTORY
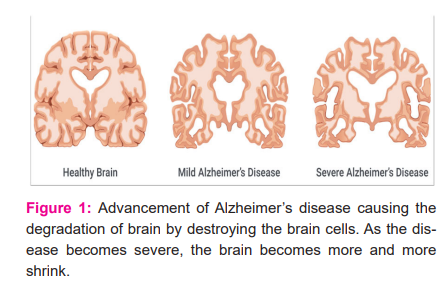
In 1906, German psychiatrist and pathologist Dr. Alois Alzheimer noticed the changes in brain tissues of a female patient who died of irrefutable mental illness and reported the first case of the disease named after him. In next five years, eleven new cases of the same illness were reported just using the terminology Alzheimer’s Disease.11 The name Senile Dementia of Alzheimer’s type(SDAT) was initially used to describe AD in individuals aged 65 years and well above, On the contrary, Classical Alzheimer's disease is used to describe patients who were younger. As time passed, the term Alzheimer's disease was used in medical literature to describe people of every age with a consistent pattern and neuropathology of typical symptoms. Nearly 47 million individuals worldwide were impacted by dementia in 2015, with a forecasted 75 million in 2030, and 131 million in 2050.13 The annual patient count is estimated at 4.6 million cases globally, which is identified as one new case every seven seconds. Figure 1.1 shows the progression of AD.
GENETICS
Alzheimer's disease is acquired in about 2% of instances in progenies (autosomal dominant). Early-onset familial Alzheimer's disease is a kind of Alzheimer's disease that starts early and progresses quickly.18Early-onset Alzheimer's disease, known as younger-onset Alzheimer's or early-onset AD, is Alzheimer's disease that develops before completing age 65. It is a rare type of Alzheimer's disease, accounting for only around 5–10% of all the cases of Alzheimer's.19 Roughly 60% have a favorable family history and 13% are autosomally dominated by AD.20 The majority of occurrences of early-onset Alzheimer's, on contrary, exhibit the same characteristics as the "late-onset" type and are not generated by genetic mutations. Early-onset familial AD can be directly linked to mutations in one of three genes which are named as an amyloid-beta precursor protein (APP) and presenilins PSEN1 and PSEN2.44 The majority of APP and presenilin gene mutations increase the formation of amyloid beta (Aβ)42, a tiny protein that is the principal component of amyloid plaques. Some mutations just change the ratio between Aβ42 and the other main forms, specifically Aβ40, without enhancing Aβ42 levels. ABCA7 and SORL-1 are two more genes linked to autosomal dominant AD.2,4,15,21
The majority of Alzheimer's cases are not inherited and are termed to as sporadic Alzheimer's disease, in which environmental and genetic conditions play an important role. In contrast to familial Alzheimer's disease, the majority of sporadic Alzheimer's disease (AD) cases develop after the age of 65.18 The start of sporadic Alzheimer's disease is delayed in fewer than 5% of instances. APOEε4 is the most powerful genetic risk factor for sporadic AD.22 APOEε4 is one of the four apolipoprotein E alleles (APOE). The ε4 allele affects the activity of APOE in lipid binding proteins in lipoprotein particles.16,22 Between 40 and 80% of persons with Alzheimer's disease have at least one APOEε4 allele.22,44 several alleles in the genome elevates the threat of AD. APOE, which codes for the lipid carrier protein apolipoprotein E (ApoE). The ApoE-4 allele increases the risk of AD thrice. Although the population is just 25%, they represent half of all Alzheimer's cases.10
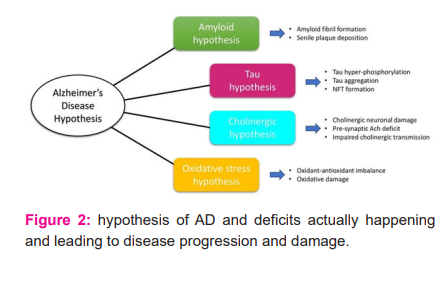
HYPOTHESIS RELATED TO ALZHEIMER’S DISEASE
Amyloid Hypothesis
For more than 25 years, the amyloid hypothesis (also known as the amyloid cascade hypothesis, the Aβ hypothesis, and so on) has been the leading explanation for the neurophysiology of Alzheimer's disease.16 The most direct anti- Aβ therapeutic technique is to limit Aβ production by attacking β - and γ -secretase.17 The original amyloid cascade theory said that “Aβ is the causative agent in Alzheimer's Disease pathogenesis, and that neurofibrillary tangles, cell death, vascular damage, and dementia come up as a direct outcome of its deposition”.23 Despite the fact that the majority of data still supports Aβ as the key initiator of the complicated pathogenic cascade in AD, more and more evidences show that Aβ serves as a trigger in the early disease process and seems to be required but not sufficient in the late stage of AD.25
Tau Hypothesis
Tau is found in neurofibrillary tangles, which are another intracellular characteristic of AD. Tau aggregation in pathological situations will damage neuron axons, resulting in neurodegeneration.40Phosphorylation, arginine monomethylation, lysine acetylation, lysine monomethylation, lysine dimethylation, lysine ubiquitylation, and serine are all forms of tau modifications.26 Tau-targeting medicines remain difficult to develop due to an insufficient information of Alzheimer's disease, a lack of reliable and specific biomarkers for diagnosis and response monitoring, and a restriction of the blood-brain barrier.27
Cholinergic Hypothesis
As the Neurochemistry involves study of the brain and neurotransmitters. The primary neurotransmitter deficit in AD is acetylcholine.28Neuronal loss causes cholinergic insufficiency that give cholinergic innervation to the cerebral cortex, notably those in the basal forebrain (the nucleus basalis of Meynert). Central cholinergic antagonists, such as atropine, can induce disorientation, which resembles AD dementia. This "cholinergic hypothesis" asserts, deficiency of ACh causes AD symptoms. It is essential to mention that even if "cholinergic deficiency syndrome" is equivalent to Parkinson's disease, the situation in AD is significantly more complex.31 Cortical and hippocampal targets that receive cholinergic input are destroyed, as are other neurotransmitter systems such as glutamate, 5-HT, and neuropeptides. To treat this, the cholinergic hypothesis was initially investigated in AD therapy with cholinesterase inhibitors. The first anti-AD medication accessible in the clinic was Tacrine, a cholinesterase inhibitor.30
SIGNS AND SYMPTOMS
Alzheimer's disease symptoms develop gradually over time. Anyone whose symptoms worsen fast should consult a doctor in order to treat AD. There might be causes for the worsening of symptoms that can be addressed. Generally, symptoms of AD are described in 3 stages- first stage (early symptoms), Middle stage (mild or moderate symptoms) and late stage (severe symptoms).
Early Stage
Memory lapses are the primary sign of AD in its early stages.For example, someone with early AD may forget recent conversations or events, misplace items, may fail to remember the names of place and objects, have difficulty thinking of the right word, ask questions repeatedly, exhibit terrible judgment or having difficulty in making decisions, and become less flexible and more hesitant to try new things.54The progressive deterioration of learning and memory in patients with AD finally leads to a confirmed diagnosis. Language, executive function, perception (agnosia), and movement execution (apraxia) impairments are more common in a small number of people than memory impairments.56Not all the memory capability is adversely impaired by AD. Episodic memories, semantic memory, and implicit memory are less altered than fresh facts or memories.55
Middle Stage
Memory impairments will worsen as Alzheimer's illness progresses. Additional symptoms such as increased confusion and disorientation may develop – for example, getting lost or roaming and not knowing the time, obsessive, repetitive, or impulsive behavior, delusions or feeling paranoid and suspiciousness about caregivers or family members, problems associated with language or speech (aphasia), disturbed sleep, mood changes (hallucinations).54 Progressive deterioration finally inhibits independence, with patients unable to do the majority of everyday tasks. Speech issues emerge as a result of a difficulty to retain language, resulting in frequent erroneous word replacements (paraphasias). Reading and writing abilities are also deteriorating.55 As time passes and AD worsens, convoluted motor sequences are less synchronized, increasing the risk of falling. Long-term memory, which was initially robust, begins to deteriorate.56

Late stage
This is the last and the most severe stage of AD. The symptoms of AD get progressively severe in the latter stages, which can be frustrating for the person with the disease, as well as their caregivers, friends, and family. Hallucinations and delusions may appear and disappear during the disease, but they might worsen as the condition advances. People suffering from AD might become aggressive, demanding, and distrustful of those around them at times.55 People in the latter phases of AD may require full-time care as well as aid with eating, moving, and personal care.54 Language is limited to basic sentences or even single words, eventually leading to total aphasia. Although aggression may persist, excessive apathy and tiredness are far more prevalent symptoms. People suffering from AD will eventually be unable to execute even the most rudimentary duties independently; their muscular mass and mobility will decrease to the extent that they are clothed and cannot feed themselves. The cause of mortality is frequently an external issue, such as pressure ulcer infection or pneumonia, rather than the disease itself.55,56
TREATMENT
AD is currently an untreatable disease. Currently, therapy plans for AD rely on symptomatic relief, with no focus on improving the target molecules. Effects and symptoms of AD can be slowed but cannot be eradicated completely. For the cure of AD, Firstly, cholinesterase antagonists such as Donepezil, Rivastigmine and Galantamine are used. Secondly, NDMA drug-like Memantine is given in the treatment of AD. Combination therapy is usually preferred for increased efficacy and relief.
Cognitive symptoms treatment
AD treatment relies on the optimization of cholinergic transmission. Tacrine, the first medication approved to treat AD, is currently being used relatively rarely due to its many side effects. Reversible cholinesterase antagonists (Catalysts for cleavage of acetylcholine into choline as well as acetate in the synaptic cleft) block cholinergic neurotransmission. For mild to severe AD, cholinesterase inhibitors are the first-line treatment.32 Lewy body dementia and vascular dementia are both treatable with cholinergic inhibitors.
Galantamine, a cholinesterase inhibitor, which has a dual form action mechanism. Aricept is the brand name for Galantamine, which was approved in 1996. It is acetylcholine esterase’s reversible inhibitor that enhances the intrinsic effect of acetylcholine on the nicotinic receptor, resulting in enhanced cholinergic neurotransmission into CNS.56 Galantamine is centrally and peripherally operating inhibitor which inhibit acetylcholinesterase in the muscles and the brain, boosting cholinergic tone. Galantamine operates as a positive allosteric modulator in neurons for nicotinic acetylcholine receptors. It is usually given in the forms of tablets or disintegrating tablets. In clinical tests of AD, Galantamine, on the other hand, encouraged improvements in cognition, global function, activities of daily living, and behavioral symptoms.53
NAMENDA is a drug used in combination with cholinesterase inhibitors to treat AD. Alzheimer's and Parkinson's are treated with it as well. N-methyl-D-aspartate receptor antagonists like Memantine are non-competitive drugs. It binds to the Mg2+ binding site on the channel, limiting activation without causing harm. Clinical deterioration is greatly slowed by the use of memantine.5,33 The drug's genuine disease-modifying effect, reduced excitotoxicity, or clinical effects are all uncertain. Headaches and dizziness are the only serious effects of using memantine.5Behavioral and psychological symptoms of dementia (BPSD), particularly during the late phase of the disease, are not uncommon in dementia. Pharmacological and non-pharmacological options should be used concurrently. They have a minor influence, and they leave many symptoms untreated, such as agitation.34 Alzheimer's patients are frequently taking medications, therefore additional treatment choices are necessary when behavioral indications develop.51
Use of a Selective serotonin reuptake inhibitors (SSRI) or an atypical antipsychotic is frequently used to treat behavioral disorders.35 The effects of these drugs on AD pathology are not well-documented. Studies asserts that stimulants have a short-term influence on cognitive functions and behavioral manifestations. Treatment delay may be implemented on a case-by-case manner. Most of the current clinical research focuses on the synergistic benefits of inhibiting cholinesterase with alterations to specific cholinergic receptors.39
Agonists for Muscarinic Receptors
Mucosal agonists are prescribed for xerostomia, urinary bladder problems, and bronchial hyperreactivity.41 Glaucoma and miotic drugs are commonly used to treat it. The involvement of cognition in muscarinic receptors is growing. Cognitive impairment induced by AD has long been treated with M1 agonists. Many other receptor subtypes appear to be engaged in the modulation of cognitive function, at least in animal models.42 Other mechanisms for selectively activating specific muscarinic receptor subtypes, such as allosteric agonists and positive allosteric modulators (PAMs), have been researched because of the absence of efficacy and substantial peripheral side effects of currently available muscarinic agonists.43 Schizophrenic and substance abuse problems as well as pain control medications all benefit from selective muscarinic subtype activators.
An example is Xanomeline, a muscarinic agonist that possesses Muscarinic-1 and Muscarinic-4 subtype selectivity and was being investigated for AD as well as schizophrenia. Anticholinesterase drugs inhibit the AchE enzyme. Cholinergic nerve terminals collect ACh and hence can elicit symptoms in the PNS and CNS that are similar to excessive cholinergic receptor stimulation.44 Many non-cholinergic toxins have also been widely used as toxicants, including agricultural pesticides, herbicides, and chemical warfare "nerve agents."
Other therapeutic strategies for treatment
While no medicine has been proved to preserve neurons, there are two potential conceptual approaches to the therapy of AD. Firstly, Treatment that helps prevent the onset of the disease by isolating the primary progenitors or targets and reduces secondary pathologies of the disease, retards disease progression or postpones disease onset, leads to the cessation or even repair of neuronal damage after disease onset, and ultimately prevents the development of AD is one approach; secondary approach is symptomatic treatment. Neurotrophins, antioxidants, statins, non-steroidal anti-inflammatory drugs (NSAIDs), hormone replacement treatment, excitotoxicity blocking, vaccination testings, immunotherapy, and secretase effectors, 7-Methoxytacrine have all been researched, but their usage remains disputed. As a result, greater research into preventative and disease-modifying therapy options is required for the elimination of AD in the general population. Beside cognitive symptoms treatments and use of muscarinic agonists other therapeutic strategies are being researched.
Use of antioxidants
Melatonin, it is a hormone derived from mammals that is primarily synthesized in the pineal gland. It collects O2 and N2-based reactants produced in mitochondria by increasing the production &activity of Glutathione peroxidase, Superoxide dismutase, and NO synthetase, and it also contributes to the diminution of oxidative damage in cells.56 In currently undergoing researches, antioxidant melatonin has been demonstrated to prevent Aβ-induced toxicity and ameliorate tau hyperphosphorylation.57 Melatonin improved the learning and memory impairments present in an APP695 transgenic mouse model through in vivo, and also inhibited Aβ-induced apoptosis in AD cell models such as mouse microglial BV-2 cells, rat astroglioma C-6 cells, and PC-12 cells through in-vitro.58 In another investigation, melatonin reduced NADPH oxidase phosphorylation via a PI3K/Akt-dependent signaling pathway in microglia vulnerable to Aβ1–42.57 According to some research, melatonin reduced Aβ burden in juvenile APP Tg2576 mice models but had zero effects on F2-IsoPs or Aβburden in older plaque-bearing mice.56
Selegiline (L-deprenyl) is a monoamine oxidase-B inhibitor with antioxidant effects that can be used to treat neurodegenerative disorders. It has the ability to rapidly produce the powerful vasodilator nitric oxide, notably in cerebral blood vessels.51 It may also protect the vascular endothelium from the harmful effects of Aβ peptide and improve the function or survival of nigral neurons by blocking oxidative deamination.52 Sano et al. demonstrated in 1997 that therapy with Selegiline (10 mg/day) decreases neuronal destruction and delays the course of AD in patients having moderately severe impairment.37 These data imply that the administration of Selegiline may postpone clinically significant functional impairment in AD patients.
DISCUSSION
1)AD, the most common form of dementia, has a multifactorial etiology, and the current therapy (AChEIs and memantine) cannot interrupt its progress and fatal outcome. This is reflected in the research programs oriented toward the development of new therapeutics able to operate on multiple targets involved in the disease progression.
2) The patents from 2016 to the present regarding the use of AChEIs in AD concern the development of new AChEIs, multitarget or multifunctional ligands, or the associations of AChEIs with other compounds acting on different targets involved in the AD.
3)The development of new multitarget AChEIs promises to identify compounds with significant therapeutic potential. However, it requires more time and effort to obtain drugs with the optimal pharmacodynamic profile. Otherwise, the research on new combinations of existing drugs with known pharmacodynamic and ADME profiles could shorten the time and reduce the costs of developing a new AD treatment. From the analyzed data, it seems more likely that a response to the urgent need to develop effective treatments for AD therapy could come more quickly from studies on drug combinations than from the development of new AChEIs.
CONCLUSION
Studies reveal that the cause of AD as a neurodegenerative condition includes extracellular amyloid plaques, intracellular neurofibrillary tangles, synaptic degradation, and neuronal death. 70% of AD risk at any given age is due to heredity. Apo lipoprotein E is the most general genetic risk factor for AD (ApoE). Aside from the genetic and biochemical aspects, a deficiency in vitamin D seems another cause of AD. In addition, brain glucose metabolism decreases in AD, which results in diabetes. Unfortunately, the currently available medications for therapy (AChEIs and memantine) only target symptoms and not the underlying cause of the condition. As a result, the possibility of new drugs that operate at the origin of the disease process and can block the gradual buildup of Aβ has been raised. It has been noted that several non-targeted therapies, such as anti-inflammatory treatment, metal chelation, antioxidant supplements, and epigenetic alterations, are more damaging than beneficial, making it impossible to predict if their correct use would enhance clinical outcomes or not. To summarize, stem cell therapy and biomarkers could be new strategies in the early diagnosis and treatment of AD. Several potential clinical trials are now undertaken, which may give new diagnostic targets and support in the resolving of the difficult AD issue.
ACKNOWLEDGMENTS
Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to authors/editors / publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed."
SOURCE OF FUNDING
The authors declare that no funding was provided for this editorial.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHORS CONTRIBUTION
Shubham S Bagade (corresponding author)
-
-
Collected the data and performed the analysis of the data.
-
Wrote the entire manuscript
-
Contacting with the journal for manuscript publication.
-
designed the model and the computational framework and analysed the data.
-
Drafted the manuscript and designed the figures.
Laxmikant B. Borse
-
Contributed in data collection and analysis.
-
Collecting key points from different journals and books
-
Directing the project.
Atul R. Bendale
-
Convinced and designed the analysis of the data
-
Working on task to keep a plagiarism-free manuscript
-
designed and directed the project
Anil G. Jadhav
-
contributed to the final version of the manuscript and supervised the project.
All authors provided critical feedback and helped shape the research, analysis and manuscript.
References:
1. Kumar K, Kumar A, Keegan RM, Deshmukh R. Recent advances in the neurobiology and neuropharmacology of Alzheimer’s disease. Biomed Pharmacother. 2018;98:297–307.
2. Gilles C, Ertlé S. Pharmacological models in Alzheimer’s disease research. Dialogues Clin Neurosci. 2000;2(3):247–55.
3. Massoud F, Gauthier S. Update on the pharmacological treatment of Alzheimer’s disease. Curr Neuropharmacol. 2010;8(1):69–80.
4. Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology. 2014;76 Pt A:27–50.
5. Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev. 2003 Autumn;9(3):275–308.
6. Webber KM, Raina AK, Marlatt MW, Zhu X, Prat MI, Morelli L, et al. The cell cycle in Alzheimer disease: a unique target for neuropharmacology. Mech Ageing Dev. 2005;126(10):1019–25.
7. Drukarch B, Leysen JE, Stoof JC. Further analysis of the neuropharmacological profile of 9-amino-1,2,3,4-tetrahydroacridine (THA), an alleged drug for the treatment of Alzheimer’s disease. Life Sci. 1988;42(9):1011–7.
8. Nehlig A. The neuroprotective effects of cocoa flavanol and its influence on cognitive performance: Cocoa flavanol and cognition. Br J Clin Pharmacol. 2013;75(3):716–27.
9. Sinyor B, Mineo J, Ochner C. Alzheimer’s disease, inflammation, and the role of antioxidants. J Alzheimers Dis Rep. 91 Int J Cur Res Rev | Vol 14 • Issue 02 • January 2022 Bagade et al: A neuropharmacological review of alzheimer’s disease 2020;4(1):175–83.
10. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17(3):327–406.
11. Wikipedia contributors. Alzheimer’s disease [Internet]. Wikipedia, The Free Encyclopedia. 2021 [cited 2021 Sep 30]. Available from: https://en.wikipedia.org/w/index. php?title=Alzheimer%27s_disease&oldid=1046539705
12. Amaducci LA, Rocca WA, Schoenberg BS. Origin of the distinction between Alzheimer’s disease and senile dementia: how history can clarify nosology. Neurology. 1986;36(11):1497–9.
13. Alzint.org. [cited 2021 Sep 30]. Available from: https://www. alzint.org/u/WorldAlzheimerReport2015.pdf
14. Hosseini L, Mahmoudi J, Pashazadeh F, Salehi-Pourmehr H, Sadigh-Eteghad S. Protective effects of nicotinamide adenine dinucleotide and related precursors in Alzheimer’s disease: A systematic review of preclinical studies. J Mol Neurosci. 2021;71(7):1425–35.
15. Canadian study of health and aging: study methods and prevalence of dementia. CMAJ. 1994;150(6):899–913.
16. Du X, Wang X, Geng M. Alzheimer’s disease hypothesis and related therapies. Transl Neurodegener. 2018;7:2.
17. Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, et al. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2(9):835–41.
18. Long JM, Holtzman DM. Alzheimer disease: An update on pathobiology and treatment strategies. Cell. 2019;179(2):312– 39.
19. Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med [Internet]. 2021;13(583). Available from: http://dx.doi.org/10.1126/scitranslmed.aaz4564
20. Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403.
21. Kim JH. Genetics of Alzheimer’s disease. Dement Neurocognitive Disord. 2018;17(4):131–6.
22. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103(15):5644–51.
23. Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5.
24. Wang J, Gu BJ, Masters CL, Wang Y-J. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612–23.
25. Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and “wingmen.” Nat Neurosci. 2015;18(6):800–6.
26. Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med. 2016;8(338):338ra66- 338ra66.
27. Chun W, Johnson GVW. The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci. 2007;12(1):733– 56.
28. Hasselmo ME, Anderson BP, Bower JM. Cholinergic modulation of cortical associative memory function. J Neurophysiol. 1992;67(5):1230–46.
29. Fine A, Hoyle C, Maclean CJ, Levatte TL, Baker HF, Ridley RM. Learning impairments following injection of a selective cholinergic immunotoxin, ME20.4 IgG-saporin, into the basal nucleus of Meynert in monkeys. Neuroscience. 1997;81(2):331–43.
30. Summers WK, Viesselman JO, Marsh GM, Candelora K. Use of THA in treatment of Alzheimer-like dementia: pilot study in twelve patients. Biol Psychiatry. 1981;16(2):145–53.
31. Fotiou DF, Stergiou V, Tsiptsios D, Lithari C, Nakou M, Karlovasitou A. Cholinergic deficiency in Alzheimer’s and Parkinson’s disease: evaluation with pupillometry. Int J Psychophysiol. 2009;73(2):143–9.
32. Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;(1):CD005593.
33. Marum R. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2009;5:237.
34. Howard RJ, Juszczak E, Ballard CG, Bentham P, Brown RG, Bullock R, et al. Donepezil for the treatment of agitation in Alzheimer’s disease. N Engl J Med. 2007;357(14):1382–92.
35. Schneider LS, Tariot PN, Dagerman KS, Davis SM, Hsiao JK, Ismail MS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525–38.
36. Bentham P, Gray R, Sellwood E, Raftery J, Rosler M, Selai CE, et al. Effectiveness of rivastigmine in Alzheimer’s disease. BMJ. 1999;319(7210):640–640.
37. Brucki SMD. Does prevention for Alzheimer’s disease exist? Dement Neuropsychol. 2009;3(3):209–13.
38. Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1(1):103– 16.
39. Chan A, Paskavitz J, Remington R, Rasmussen S, Shea TB. Efficacy of a vitamin/nutriceutical formulation for early-stage Alzheimer’s disease: a 1-year, open-label pilot study with a 16-month caregiver extension. Am J Alzheimers Dis Other Demen. 2008;23(6):571–85.
40. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3(4):519–26.
41. James PK, Christopher MF, Proneness to psychological distress is associated with risk of Alzheimer’s disease, Neurology Sep 2004, 63 (5) 941; DOI: 10.1212/WNL.63.5.941
42. Evans, DA., Funkenstein HH., Albert MS, Scherr PA., Cook NR., Chown MJ et al. (1989). Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. JAMA, 262(18), 2551–2556.
43. Mahley RW, Weisgraber KH, & Huang Y. (2006). Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America, 103(15), 5644–5651. https://doi.org/10.1073/pnas.0600549103 44. Selkoe DJ & Podlisny MB. (2002). Deciphering the genetic basis of Alzheimer’s disease. Annual review of genomics and human genetics, 3, 67–99. https://doi.org/10.1146/annurev. genom.3.022502.103022
45. Ohnishi A, Mihara M, Kamakura H., Tomono Y, Hasegawa J, Yamazaki Ket al. (1993). Comparison of the pharmacokinetics of E2020, a new compound for Alzheimer’s disease, in healthy young and elderly subjects. J. Clin. Pharmacol. 33(11), 1086– 1091. https://doi.org/10.1002/j.1552-4604.1993.tb01945.x 46. Scott LJ & Goa KL. (2000). Galantamine: a review of its use in Alzheimer’s disease. Drugs, 60(5), 1095–1122. https://doi. org/10.2165/00003495-200060050-00008
47. Hossain M, Jhee SS, Shiovitz T, McDonald C, Sedek G,Pommier Fet al.(2002). Estimation of the absolute bioavailability of rivastigmine in patients with mild to moderate dementia of the Alzheimer’s type. Clinical pharmacokinetics, 41(3), 225–234. https://doi.org/10.2165/00003088-200241030-00006
48. Dooley M, & Lamb HM. (2000). Donepezil: a review of its use in Alzheimer’s disease. Drugs & aging, 16(3), 199–226. https:// doi.org/10.2165/00002512-200016030-00005
49. Pamila EP. Current investigational drugs For treatment of Alzheimer’s disease, Frontiers in Clinical Drug Research - Alzheimer Disorders, Volume 4, 2015, 176-235.
50. Bhana N, Perry CM. Olanzapine: a review of its use in the treatment of bipolar I disorder. CNS Drugs. 2001 ;15(11):871-904. DOI: 10.2165/00023210-200115110-00005.
51. Bostwick JR, Guthrie SK, Ellingrod VL: Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009 Jan;29(1):64-73. doi: 10.1592/phco.29.1.64. (PubMed ID 19113797)
52. Christian C. Felder, Chapter One - GPCR drug discovery-moving beyond the orthosteric to the allosteric domain, Advances in Pharmacology, Academic Press, Volume 86, 2019, Pages 1-20, ISSN 1054-3589.
53. Lilienfeld S. (2002). Galantamine--a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS drug reviews, 8(2), 159–176. https://doi.org/10.1111/j.1527-3458.2002.tb00221.x
54. Carlesimo GA, Oscar-Berman M (June 1992). “Memory deficits in Alzheimer’s patients: a comprehensive review”. Neuropsychology Review. 3 (2): 119–69. doi:10.1007/BF01108841. PMID 1300219. S2CID 19548915
55. Jelicic M, Bonebakker AE, Bonke B (1995). “Implicit memory performance of patients with Alzheimer’s disease: a brief review”. International Psychogeriatrics. 7 (3): 385–92. doi:10.1017/S1041610295002134. PMID 8821346.
56. Förstl H, Kurz A (1999). “Clinical features of Alzheimer’s disease”. European Archives of Psychiatry and Clinical Neuroscience. 249 (6): 288–90. doi:10.1007/s004060050101. PMID 10653284. S2CID 26142779
57. Zhou J, Zhang S, Zhao X, & Wei T. (2008). Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-beta1-42. J. pineal research, 45(2), 157–165. https://doi.org/10.1111/j.1600- 079X.2008.00570.x
58. Feng Z, & Zhang JT. (2004). Protective effect of melatonin on beta-amyloid-induced apoptosis in rat astroglioma C6 cells and its mechanism. Free radical biology & medicine, 37(11), 1790– 1801. https://doi.org/10.1016/j.freeradbiomed.2004.08.023
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





