IJCRR - 7(19), October, 2015
Pages: 11-15
Date of Publication: 10-Oct-2015
Print Article
Download XML Download PDF
FUNGAL INFECTION COMPLICATING A CASE OF PULMONARY ALVEOLAR PROTEINOSIS
TO FATAL OUTCOME
Author: Narendra Kumar Narahari, Bhaskar Kakarla, Shantveer G. Uppin, Harsha Vardhana K.R., Mohammed Ismail Nizami, Rajendra Prasad Boddula
Category: Healthcare
Abstract:Aim: To present clinico-pathological findings in a case of pulmonary alveolar proteinosis complicated by fungal infection. Case Report: A 48-year-old female presented with progressively increasing exertional breathlessness for the past 6 months. On imaging she showed extensive air space filling with crazy paving pattern in the left lung with multiple nodular lesions in the right lung and mild bilateral pleural effusions. Core needle biopsies of the lung showed features consistent with pulmonary alveolar proteinosis in the left lung and fungal infection in the right lung. Discussion: Infections with unusual organisms can complicate pulmonary alveolar proteinosis (PAP) due to inherent alveolar macrophage dysfunction and intra alveolar accumulation of surfactant offering a good culture medium for the microbes. The risk is further increased if such patients are treated with steroids. Conclusion: Opportunistic infections can complicate the clinical course of PAP which is associated with high mortality. High index of suspicion, early diagnosis and aggressive treatment can prevent the adverse outcomes
Keywords: Pulmonary alveolar proteinosis, Aspergillus, Fungal infection, Steroid therapy
Full Text:
INTRODUCTION
Pulmonary alveolar proteinosis (PAP) is a very rare entity characterized by accumulation of lipoproteinacious material in the alveoli which impairs gas exchange. [1] The incidence of PAP in the world is estimated to be around 0.2 cases per million. [1] To the best of our knowledge, not more than 13 cases were reported in Indian literature till now. [2,3,4,5] Infections with unusual organisms can occur in PAP with lungs being the most common site of infection. The alveolar macrophage dysfunction and intra alveolar accumulation of surfactant in these patients offers a good culture medium for the microbes.[6,7] Literature showed that approximately 5 to 13% of patients with autoimmune PAP present with these opportunistic infections. [7,8] These infections either precede or succeed the clinical course of PAP. The overall survival rate of PAP is very poor when complicated by these infections and the highest mortality is seen with fungal infections. [9] Here we present one such case of PAP complicated with fungal infection.
CASE HISTORY
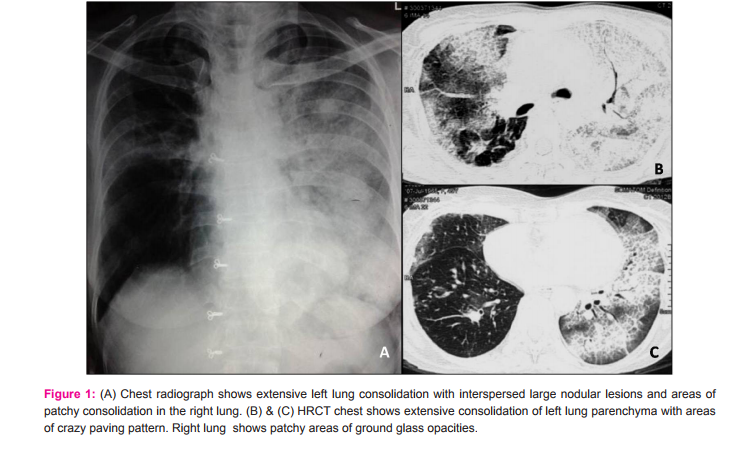
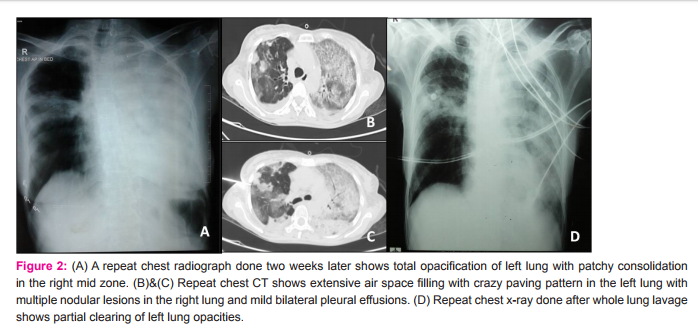
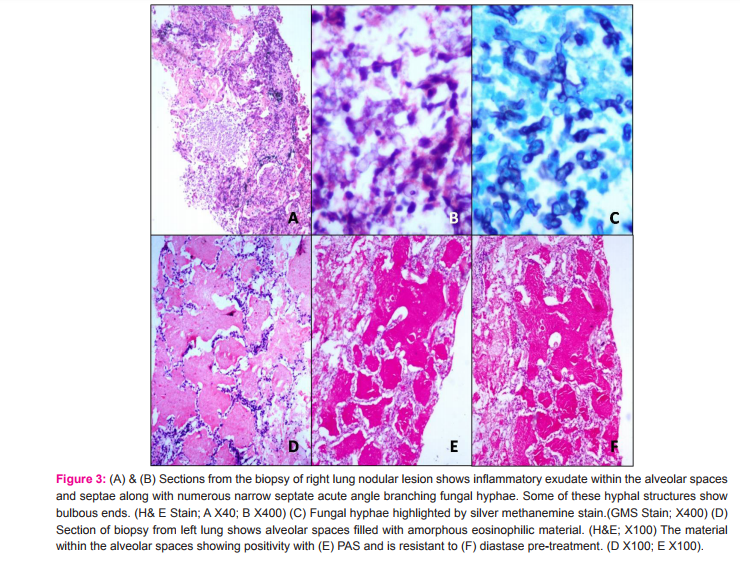
A 48 year old female presented to us with progressively increasing exertional breathlessness for the past 6 months, with breathlessness at rest since the past 2 months. She was complaining of loss of appetite for the past 4 months. There were no complaints of fever, cough and hemoptysis. She was a non-smoker and there was no significiant history of environmental and occupational exposure. She was not a known diabetic or hypertensive. She was diagnosed with sputum negative pulmonary tuberculosis 2 years back and received anti tubercular therapy for 6 months. Before presenting to us, she was admitted in a local private hospital for similar complaints. Her chest radiograph showed extensive left lung consolidation with interspersed large nodular lesions and areas of patchy consolidation in the right lung (Fig.1A). High resolution computed tomography (HRCT) of chest showed extensive consolidation of left lung parenchyma with areas of crazy paving pattern. Right lung also showed patchy areas of ground glass opacities (Fig.1B and C). Based on these clinical and radiological findings, she was treated outside with antibiotics, steroids and supported with non invasive ventilation for 10 days duration and later referred to us in view of persisting respiratory distress. The patient was thin built and ill-nourished. Routine blood investigations were within normal limits. Chest auscultation revealed bilateral lung crepitations. Her arterial blood gas analysis showed hypoxia with type I respiratory failure. Viral markers for HIV and HBsAg were negative. Sputum examination was negative for acid fast bacilli and fungal elements. A repeat chest radiograph at our institute showed total opacification of left lung with patchy consolidation in the right mid zone (Fig.2A). Repeat chest CT showed extensive air space filling with crazy paving pattern in the left lung with multiple nodular lesions in the right lung and mild bilateral pleural effusions. (Fig.2B and C) In view of the clinical and radiological findings and persisting lung lesions, a differential diagnosis of pulmonary alveolar proteinosis or invasive mucinous adenocarcinoma of lung was considered. CT guided biopsy was obtained from both lung lesions on separate occasions due to varied morphology of the lesions in either lung. Sections from the biopsy obtained from right lung nodular lesion showed inflammatory exudate within the alveolar spaces and septae along with numerous narrow septate acute angle branching fungal hyphae. Some of these hyphal structures showed bulbous ends. These fungal hyphae were highlighted by silver methanemine stain. (Fig.3A-C) These features were consistent with invasive fungal infection possibly due to Aspergillus species. Biopsy obtained from left lung showed alveolar spaces filled with amorphous eosinophilic material. This material was PAS positive and diastase resistant. These findings were consistent with PAP. (Fig.3D-F) In view of above pathological findings patient was started on voricanazole. Her general condition did not improve and respiratory distress progressed in spite of antifungals and non invasive ventilatory support. Whole lung lavage (WLL) of left lung was done with 8 liters of saline under general anesthesia with double lumen endotracheal tube intubation, in view of her persisting respiratory distress. Her saturations improved and chest radiograph showed partial clearing of left lung opacities (Fig.2D). Further sequential lavage of the lung was planned, but she succumbed to sepsis with multiorgan failure.
DISCUSSION:
Three main categories of PAP are defined based on etiology. (i) Autoimmune/idiopathic PAP accounts for 90% of the cases and is characterized by loss of GM-CSF signaling due to presence of neutralizing GM-CSF auto antibodies. This leads to a state of functional deficiency which interferes with surfactant clearance mechanisms. [10] (ii) Secondary PAP may develop with chronic infections, hematological disorders, inhalation of dust and fumes and immunodeficiency disorders. In secondary PAP, there is acquired loss of GM–CSF signaling due to reduction in number or certain functions of alveolar macrophages. (iii) Congenital or genetic PAP is a autosomal recessive disease occurring in children and exhibiting a wide range of phenotypic variations. [6] PAP has a variable clinical course and presentation which can range from spontaneous resolution to progressive worsening. [11] Most of the patients present with progressive dyspnea on exertion although few of them are asymptomatic. Fever and hemoptysis are rare if present should suggest secondary infection. [6,10] Chest radiograph is often inconclusive which reveals bilateral symmetric, patchy, peri - hilar alveolar opacities without air bronchogram resembling pulmonary edema. [1] Chest CT is more diagnostic showing typical geographic pattern of ground glassing with superimposed septal reticulations forming a crazy paving pattern. Although the CT finding of crazy-paving is highly characteristic of PAP, it is also seen in many other conditions including left heart failure, pneumonia (especially pneumocystis pneumonia), alveolar hemorrhage, bronchoalveolar carcinoma, lymphangitic carcinomatosis, diffuse alveolar damage (adult respiratory distress syndrome), radiation or drug induced pneumonitis, hypersensitivity pneumonitis, and pulmonary veno-occlusive disease. Mediastinal lymph node enlargement, pulmonary nodules, pleural effusions and focal consolidation in the lung imaging are atypical and if present should suggest an opportunistic infection or malignancy. [12,13] The presence of interspersed nodular lesions and pleural effusions in the chest CT done after the administration of steroids suggested the possibility of superadded infection in our patient which was proved to be invasive fungal infection of Aspergillus species on histopathological examination. We consider infection to be a secondary event due to synergistic effect of inherent alveolar macrophage dysfunction, rich intra alveolar accumulation of surfactant offering a good culture medium for the microbes and to the administration of steroids. Literature also suggests that most of the infections encountered in PAP are secondary to the disease process rather than a primary event, as the lavage fluid samples were found to be microbiologically sterile in many patients. [7] In the present case, the appearance of the nodules after starting steroid therapy gives more credence to the role of steroids. The major complication of PAP is infections with opportunistic and unusual organisms like aspergillus species, nocardia, mycobacterium species, pneumocystis jiroveci and viruses which are responsible for significiant mortality if not diagnosed early and treated aggressively. [6] In a recent article by Punatar et al,[9] opportunistic infections preceded (40%), occurred simultaneously (27%) or followed (33%) the diagnosis of PAP. Lungs were the most common site involved and most common opportunistic infections were mycobacterial followed by fungal and nocardial species. The overall survival rate was found to be 56%, with mycobacterial being the greatest while the fungal carrying the poor survival with high mortality. The administration of steroids in PAP is found to be highly detrimental [6] as it can exacerbate these opportunistic infections as was seen in our case. GM-CSF cytokine plays a key role in the pathophysiology of PAP. GM-CSF by inducing terminal differentiation of alveolar macrophages can cause normal surfactant clearance mechanisms and certain functions of alveolar macrophages like antigen presentation and phagocytosis. [14] Concomitant neutrophilic dysfunction due to GM- CSF antibodies, defective chemotaxis and phagocytosis due to impaired alveolar function, superoxide production and secretion of pro inflammatory cytokines predisposes PAP to opportunistic infections. [6,14] The presence of anti GM- CSF antibodies in the BAL which can differentiate between primary and secondary PAP couldn’t be demonstrated in our case due to lack of availability. Although histopathological examination of lung tissue is gold standard, the diagnosis of PAP can be made confidentially by characteristic HRCT appearance along with typical BAL findings. [15] The typical appearance of milky white opalescent, viscous material in the bronchial lavage confirms the diagnosis. Histopathology usually reveals granular lipoproteinaceous material filling the alveoli which stains deep pink with PAS stain. [7] However BAL and lung biopsy are usually done to exclude infections in case of clinical suspicion. The presence of atypical radiological findings and diagnostic dilemma in the present case prompted for more invasive procedures like lung biopsy which demonstrated concomitant fungal infection along with PAP. Therapeutic whole lung lavage is the accepted standard of treatment in PAP. WLL, apart from physical removal of proteinaceous material, can improve alveolar macrophage function by correcting the defects in phagocytosis and migration thereby decreasing the risk of opportunistic infections. [16] Sufficient accumulation of surfactant to cause progressive respiratory failure or exercise desaturation warrants WLL. Other modes of treatment are exogenous administration of GM-CSF, which by restoring the functional availability could alter the natural course of the disease. Reducing the levels of anti GM – CSF auto antibodies with plasmapheresis and rituximab are under investigation. The therapeutic efficiency of all these, when compared to WLL is found to be inferior. [7]
CONCLUSIONS
Opportunistic infections can complicate the clinical course of PAP which is associated with high mortality. High index of suspicion, early diagnosis and aggressive treatment can prevent the adverse outcomes. The presence of nodules, pleural effusions and focal consolidations in lung imaging along with crazy paving pattern should prompt for more invasive procedures for diagnosing these infections. Corticosteroids can aggravate these opportunistic infections and should not be used during the management or in the initial suspicion of PAP. [6]
ACKNOWLEDGEMENT
Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to authors / editors / publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed.
Source of funding: nil
Conflict of interest: nil
References:
1. Borie R, Danel C, Debray MP, Taille C, Dombret MC, Aubier M. Pulmonary alveolar proteinosis. Eur Respir Rev 2011; 20: 98–107.
2. Khan A, Agarwal R, Aggarwal AN, Bal A, Sen I, Yaddanapudi LN et al. Experience with treatment of pulmonary alveolar proteinosis from tertiary care centre in north India. Indian J Chest Dis Allied Sci 2012; 54:91-7.
3. Hasan A, Ram R, Swamy T. Pulmonary alveolar proteinosis due to mycophenolate and cyclosporine combination therapy in a renal transplant recipient. Lung India 2014; 31: 282-4.
4. Chaudhuri R, Prabhudesai P, Vaideeswan P, Mahashur AA. Pulmonary alveolar proteinosis with pulmonary tuberculosis. Ind. J. Tub 1996; 43: 27-9.
5. Davis KR, Vadakkan DT, Krishnakumar EV, Anas AM. Serial bronchoscopic lung lavage in pulmonary alveolar proteinosis under local anesthesia. Lung India 2015; 32: 162-4.
6. Shah PL, Hansell D, Lawson PR, Reid KB, Morgan C. Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis. Thorax 2000; 55: 66-77. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: Progress in the first 44 years. Am J Respir Crit Care Med 2002; 166: 215-35.
7. Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med 2008; 177: 752-62.
8. Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect 2012; 65: 173-9.
9. Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med 2003; 349: 2527-39.
10. Kariman K, Kylstra JA, Spock A. Pulmonary alveolar proteinosis: prospective clinical experience in 23 patients for 15 years. Lung 1984; 162: 223-31. Frazier AA, Franks TJ, Cooke EO, Mohammed TL, Pugatch RD, Galvin JR. From the archives of the AFIP: pulmonary alveolar proteinosis. Radiographics 2008; 28: 883-99.
11. Holbert JM, Costello P, Li W, Hoffman RM, Rogers RM. CT features of pulmonary alveolar proteinosis. AJR Am J Roentgenol 2001; 176: 1287-94.
12. Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. 2007; 356:567-79.
13. Bonella F, Bauer PC, Griese M, Ohshimo S, Guzman J, Costabel U. Pulmonary alveolar proteinosis: new insights from a single – center cohort of 70 patients. Respir Med 2011; 105: 1908-16.
14. Hoffman RM, Dauber JH, Rogers RM. Improvement in alveolar macrophage migration after therapeutic whole lung lavage in pulmonary alveolar proteinosis. Am Rev Respir Dis 1989; 139:1030-2.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





