IJCRR - 13(21), November, 2021
Pages: 39-41
Date of Publication: 09-Nov-2021
Print Article
Download XML Download PDF
\" Myotonic Myopathy - A Case Report \"
Author: Kakkad A., Ramanandi V., Desai A.
Category: Healthcare
Abstract:Background: Myotonia is a quite rare condition. The involvement of multiple systems makes the management of conditions difficult. The role of physiotherapy for such a rare condition is unexplored. Objective: The objective of this case study is to be clearer about the role of physiotherapy in the treatment of Myotonic-myopathy. Discussion: In this case report, the disease and its medical and physiotherapy management are discussed in brief. Conclusion: Patients suffering from myotonic myopathy can be benefited from a holistic approach with the combination of medicine and physiotherapy.
Keywords: Myotonia, Myopathy, Myotonic Myopathy, Muscle Relaxation Difficulty, Quality of life, Physiotherapy
Full Text:
Introduction
It is a rare genetic multi-system disorder of late childhood or adult-onset characterized by mild myotonia, muscle weakness, and rarely cardiac conduction disorders.1 Myotonic Dystrophy (DM) is a type of muscular dystrophy that can affect skeletal muscles and other viscera in the human body. “Myotonia can be defined as an inability to relax muscles at will.” The term “muscular dystrophy” denotes progressive degeneration of muscles, with weakness and atrophy of the muscle. Myotonic dystrophy is often written as “DM” due to the Greek name, dystrophia myotonica. Steinert disease is another name occasionally used for this.2 Other synonyms are Curschmann-Batten-Steinert syndrome, Myotonia Atrophica, Myotonic Muscular Dystrophy, Proximal Myotonic Myopathy, Ricker Syndrome. It refers to two rare genetic disorders of muscle that affect multiple systems of the body. There are two main types.DM type 1 (DM1) is classified as mild DM1, classic DM1 as well as congenital DM1. Mild DM1 is identified by clouding of the eyes lenses (cataracts) and maintained muscle contractions (myotonia), where the muscle does not relax after contraction. Classic DM1 is identified by weakness and wasting of muscle (atrophy), myotonia, early onset of cataracts (before 50 years of age), and heart conduction abnormalities of electrical impulses. Congenital DM1 is identified by the weakness of muscle (hypotonia), difficulty in breathing, disability, and early chances of death. DM type 2 (DM2) manifests similar clinical features to DM1, but it is noted generally a less severe form of disorder and does not produce congenital disease. DM1 is due to a change in the DMPK gene. DM2 is due to a change in the CNBP gene. All these changes are in an autosomal dominant manner.3 There is currently no cure for myotonic dystrophy, there are ways to help manage the condition.4 It is characterized by myotonia, progressive muscle weakness, cataracts, and cardiac abnormalities as well as frontal balding and gonadal insufficiency in males. The primary genetic abnormality responsible for myotonic dystrophy has been identified as an expanded trinucleotide repeat (CTG) in the DM gene on chromosome 19.5 The progression of DM is variable among different individuals, but generally, symptoms progress low. Life span is reduced in patients having congenital DM1 and is likely reduced in patients having childhood DM1 and classic (adult-onset) DM1.6 It is an autosomal dominant inherited disorder.7
Case Report
Permission was taken from the head of the institution. Here reported case is of 18 years old male suffering from myotonic myopathy. At the age of 16 years, the patient had started feeling difficulty in walking and difficulty standing from cross leg sitting from the floor. During investigations, Creatine Phosphokinase level was found high i.e. 1084 U/L. Motor and Sensory nerve conduction studies were found normal. Electromyography revealed small amplitude, short duration, and polyphasic Motor Unit Action Potentials for all muscles when voluntary activity was recorded along with the presence of full and early recruitment with submaximal exercises and interference pattern was recorded suggestive of myotonic discharge. Pulmonary Function Test demonstrated severe restriction at the age of 17 years. 2DEcho was found with a normal Left Ventricular Ejection Fraction. The patient was given medicines as a supportive treatment only. Medicines used were Tab. Bonwell (Calcium Carbonate + Vitamin D3 1000 IU) and Tab. Evion (Vitamin E) 200 mg and also multivitamins occasionally.
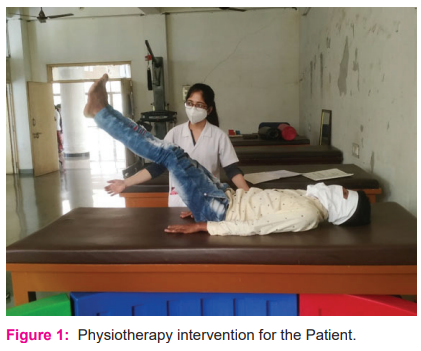
Physiotherapy assessment demonstrated an absence of all the jerks in presence of normal tone and normal sensations. Muscle power assessed by Modified Medical Research Council grading was reduced in all the muscles of limbs and trunk. Physiotherapy treatment is directed towards developing good strength in all four limbs and trunks, functional balance, and gait training with more focus on functional rehabilitation as shown in Figure 1. The patient is currently under a strengthening regimen for all four limbs and functional training for activities of daily living. The patient is currently doing all his daily living activities along with continuing his job and can deal with his personal and social responsibilities well. Improvement in cardiopulmonary endurance is a long-term goal for maintaining a longer and quality life for the patient.
Discussion
The patient represents a rare case of myotonic myopathy. The patient is selected for the study as proper medical management along with appropriate physiotherapy showed improvement in the patient’s quality of life by increasing muscle strength. There are very few researches available for the role of physiotherapy in myotonic myopathy so more numbers of researches are needed to be done for long-term duration. This can also help for creating awareness about changing the common belief that for this type of patient, only maintenance of condition and preventing complication is not sufficient. This type of patient can be given improved strength, endurance, and quality of life even though the condition itself is not curable. There is no current evidence-based standard of care for patients with myotonic myopathy and no research was found.7
Conclusion:
From the present case study, it is concluded that patients suffering from myotonic myopathy can be benefited from a holistic approach with a combination of medicine and physiotherapy. The patient’s functional independence and quality of life can be improved by physiotherapy intervention. Future research is required for more pieces of evidence of physiotherapy in the treatment of Myotonic Myopathy.
Declaration by Patient:
Informed written consent was signed by the patient.
Financial Support & Sponsorship:
Nil
Conflict of Interest:
None
Acknowledgement:
We would like to thank the Management of SPB Physiotherapy College, Surat for allowing us this research and Staff members for supporting this research.
Author’s contribution:
The first & second authors assessed and treated discussed the case and the third author reviewed the same.
Source of Funding:
Nil
References:
References
-
Reserved I. Orphanet: Proximal myotonic myopathy [Internet]. Orpha.net. 2020 [cited 6 April 2020]. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=606
-
Diseases - Myotonic Dystrophy DM - Top Level | Muscular Dystrophy Association [Internet]. Muscular Dystrophy Association. 2020 [cited 6 April 2020]. Available from: https://www.mda.org/disease/myotonic-dystrophy
-
Myotonic Dystrophy - NORD (National Organization for Rare Disorders) [Internet]. NORD (National Organization for Rare Disorders). 2020 [cited 6 April 2020]. Available from: https://rarediseases.org/rare-diseases/dystrophy-myotonic/
-
The myotonic dystrophies - Muscular Dystrophy UK [Internet]. Musculardystrophyuk.org. 2020 [cited 6 April 2020]. Available from:https://www.musculardystrophyuk.org/about-muscle-wasting-conditions/myotonic-dystrophy/myotonic-dystrophy-factsheet/
-
Stephan E. et. al. Proximal Myotonic Myopathy: Clinical, Neuropathologic, and Molecular Genetic Features. ACLSS. 2001;31(2):140-146
-
Diseases - Myotonic Dystrophy DM - Top Level | Muscular Dystrophy Association [Internet]. Muscular Dystrophy Association. 2020 [cited 6 April 2020]. Available from: https://www.mda.org/disease/myotonic-dystrophy
-
Schulte-Mattler W, Zierz S, Eger K. Proximalemyotone. Myopathie (PROMM). Der Nervenarzt. 1997;68(10):839-844.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





