IJCRR - 13(15), August, 2021
Pages: 77-80
Date of Publication: 10-Aug-2021
Print Article
Download XML Download PDF
Holoprosencephaly: Four Case Reports and Review of Literature
Author: Forci Khenata, Alami Mohamed Hassan, Bouaiti El Arbi, Mdaghri Alaoui Asmaa, Thimou Izgua Amal
Category: Healthcare
Abstract:Introduction: Holoprosencephaly (HPE) is a complex cerebral malformation due to an incomplete division of the forebrain, occurring between the 18th and the 28th day of embryonic life and affecting both the brain and the face. There are 3 increasing degrees of severity: the lobar, semi-lobar, and alobar holoprosencephaly. Case Report: We report 4 cases of holoprosencephaly identified following natural childbirth, in 75% of cases in patients of low socioeconomic level, with the first-degree consanguinity in 25% of cases. The pregnancies were not properly monitored, no folic acid supplementation had been provided and, in 25% of cases, consumption of fenugreek was reported. The antenatal diagnosis was performed in 25% of cases with a male predominance, a sex ratio at 2. Discussion: Holoprosencephaly is a complex human brain malformation resulting from incomplete cleavage of the prosencephalon. HPE is usually associated with facial abnormalities, such as cyclopia, proboscis, or cleft lip/palate in severe HPE cases. In our study, the prevalence is assessed at 0.9/10,000 births, l'HPE is associated with hexadactyly in 50% of cases and with sexual ambiguity in 25% of cases. Prenatal diagnosis is based on ultrasound and MRI. The aetiology of HPE is multifactorial, and the prognosis depends on the type of HPE and the associated facial anomalies. Conclusion: Holoprosencephaly is a complex brain malformation. For the majority of cases, the prognosis is grim. Indeed, 75% of cases were fetal death in utero (FDIU) and one case died immediate post-partum.
Keywords: Antenatal diagnosis, Cyclopia, Holoprosencephaly, Proboscis, Nodal/Hedgehog signalling, Prognosis
Full Text:
Introduction
Described for the first time by Kundrat in 1882, who used the term arhinencephaly for this entity.1 In the early 1960s, DeMyer and colleagues named the malformation holoprosencephaly.1-4
Holoprosencephaly is a complex human brain malformation resulting from incomplete cleavage of the prosencephalon into right and left hemispheres, occurring between the 18thand the 28thday of gestation. The estimated prevalence of HPE is less than 1/10,000 live births and the incidence may be as high as 1/250 in first-trimester conceptuses.5Three levels of increasing severity are described: lobar HPE, semi lobar, and alobar HPE. The middle interhemispheric variant (MIH), also known as syntelencephaly, is classified as a variant of HPE with midline interhemispheric fusion. HPE is most often associated with facial abnormalities; such as cyclopia, proboscis, Median or bilateral cleft lip and palate in severe cases, solitary ocular or median hypotelorism central incisor in minor forms. These latter midline defects can occur without the brain malformations and are then called microforms.
The aetiology of HPE is heterogeneous and complex. It can be due to cytogenetic aberrations and/or of syndromic origin, sometimes isolated in familial and sporadic forms. Loss-of-function heterozygous mutations in components and regulators of the Nodal and Hedgehog (Hh) signalling pathways are associated with HPE. Complications of HPE are characterized by developmental delay and feeding difficulties, epilepsy, instability of temperature, heart rate and breathing. Treatment is symptomatic and supportive and requires multidisciplinary management. The prognosis depends on the type of HPE and the facial anomalies associated. Severely affected children have a very poor prognosis. Child outcome depends on the HPE severity and the medical and neurological complications associated.
Case Reports:
We report four (4) cases of holoprosencephaly that are identified following natural childbirth at the “Les Orangers” Maternity and Reproductive Health Hospital of Rabat. They are listed as follows:
1st Case:(Figures: 1 & 2)
26-year-old, G1P0, and reporting the following elements:
-
Firstdegree inbreeding;
-
Intake of Fenugreek and plants during the first trimester of pregnancy;
-
No followup during pregnancy, and no supplement of iron or folic acid
-
Ultrasound performed at 27 weeks of amenorrhea (WA) by a general practitioner objectifying a hydramnios, no morphological ultrasound was performed
-
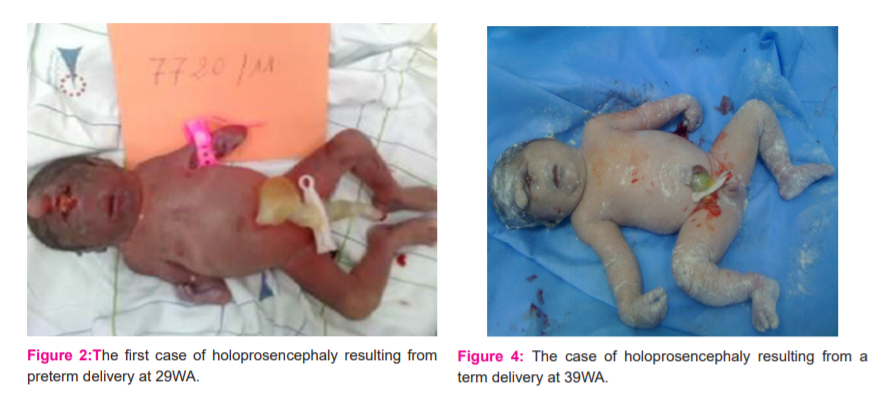
Admitted to the maternity ward in the expulsion phase at 29WA + 5 days, stillborn female, birth weight = 770 g, Apgar = 0
-
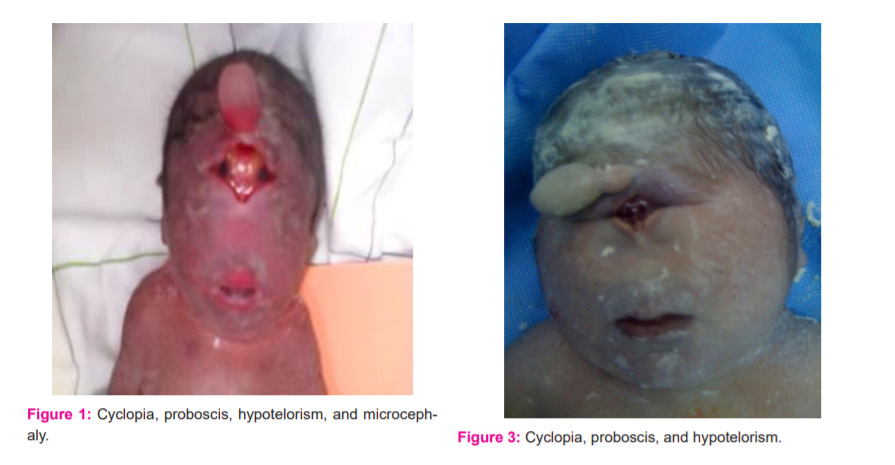
Particular aspect: the confluence of the orbits with a fusion of the eyeballs, a proboscis between the eyes with a single nostril, hypotelorism, microcephaly and sexual ambiguity.
-
A karyotype was proposed to the couple but was not performed
-
All contact was unfortunately lost with the young couple.
2nd Case:
30-year-old, G1P0, low socio-economic level, and reporting the following elements:
-
Inbreeding = 0, Diabetes = 0
-
Intake of Fenugreek and plants = 0
-
No follow-up during pregnancy, and no supplement of iron or folic acid
-
Antenatal diagnosis not performed,
-
Having consulted at 30WA with general practitioner, where the diagnosis of a hydramnios was made
-
Cytotec initiation, male stillborn, birth weight=1000g, Apgar=0
-
Clinical examination at birth = holoprosencephaly
-
Karyotype: not performed due to lack of financial means
3dCase:
42-year-old, gravida 3 para 3, average socio-economic level, and reporting the following elements:
-
Inbreeding = 0, Diabetes = 0
-
Intake of Fenugreek and plants = 0
-
Poor follow-up of pregnancy, iron-supplemented at T3, and no intake of folic acid
-
Antenatal diagnosis not performed
-
Vaginal delivery at 38WA of a newborn male, birth weight=2700g, Apgar=3 to 1 minute, apparent death, died in immediate post-partum
-
Clinical examination at birth = holoprosencephaly, with agenesis of the nose and the two eyeballs, cyclopia, hexadactyly.
4thCase: (Figures 3 & 4)
35-year-old, gravida 6 para 4, followed for high blood pressure since 32WA, low socio-economic level, and reporting the following elements:
-
Inbreeding = 0, Diabetes = 0
-
Intake of Fenugreek and plants = 0
-
Pregnancy follow-up, intake of Methyldopa 500x3/day during the third trimester
-
Antenatal diagnosis made at 26WA= objectified polymalformatif syndrome, with cyclopia, hexadactyly
-
The birth occurred at 39WA, female stillborn, hypertrophic, birth weight=2070g
-
Clinical examination at birth confirms the prenatal diagnosis.
Discussion:
In our context, the prevalence is assessed at 0.9/10,000 births. Three levels of increasing severity are described:(i) lobar HPE, where the right and left ventricles are separated, but with some continuity across the frontal cortex; (ii) semilobar HPE with a partial separation; and (iii) the most severe form, alobar HPE, with a single brain ventricle and no interhemispheric fissure. The middle interhemispheric variant (MIH), also known as syntelencephaly, is classified as a variant of HPE with midline interhemispheric fusion.HPE is usually associated with facial abnormalities, such as cyclopia, proboscis, or cleft lip/palate in severe HPE cases. In our study, HPE is associated with hexadactyly in 50% of cases and with sexual ambiguity in 25% of cases, with a male predominance, the sex ratio at 2.
The aetiology of HPE is multifactorial. Chromosomal and genetic abnormalities, teratogen exposure, and/or syndromic association have been reported in the literature. Karyotype study finds a chromosomal abnormality such as trisomy 13, in 24%–45% of all HPE cases.6While 10% of “normal” karyotype have microdeletions or duplications, heterozygous, loss-of-function mutations in components and regulators of the Nodal and hedgehog (Hh) signalling pathways are associated with HPE.7,8Ethanol9, maternal diabetes - potentially increasing the risk 200-fold, ethyl alcohol, smoking, and retinoic acid have been described as possible causes of HPE.10In our cases, we have determined the following risk factors:75% patients are of low socioeconomic level, with the first-degree consanguinity in 25% of cases. The pregnancies were not properly monitored, no folic acid supplementation was provided and, in 25% of cases, consumption of fenugreek was reported.
Prenatal diagnosis is based primarily on ultrasound and MRI in the third trimester of pregnancy, at birth on clinical, ETF, CT and especially brain MRI, and genetic diagnosis involves the karyotype. The contribution of prenatal imaging in the assessment and diagnosis of embryological abnormalities, in particular of the CNS, was demonstrated by Lekshmiet al.which classified them in category A.11Prenatal ultrasound can detect the central nervous system and facial abnormalities of severe HPE as early as the first trimester, but is less sensitive for detection of milder forms of HPE. Fetal MRI would provide better characterization of the malformations in the third trimester of the pregnancy. In our study, the prenatal diagnosis has been established in 25% of cases, i.e. the case of pregnancy followed for arterial hypertension started from 32 weeks on Methyldopa 500 three times a day.
Treatment is symptomatic and requires multidisciplinary management. The prognosis depends on the type and the severity of HPE and the medical and neurological complications associated. For the majority of cases, the prognosis is grim. Indeed, 75% of our cases were FDIU and one case died in immediate post-partum.
Conclusion
HPE is a complex developmental brain malformation commonly associated with facial anomalies, whose prognosis depends on the type of HPE and facial anomalies associated. The best diagnostic procedure is ultrasound examination, completed eventually by MRI. There are a number of causes of HPE, including genetic alterations and environmental effects. In our context, the four (4) cases represented severe forms of HPE, explaining the poor prognosis, and thus prompting the need for a systematic follow-up of any pregnancy and the implementation of an antenatal ultrasound screening program allowing the early diagnosis of these anomalies and adequate care at birth for viable forms on the one hand and on the other hand, the indication of a possible medical termination of pregnancy for lethal forms if the parents so request and legislation allow this (note that the text of the law is being revised to include in addition to the indication of the maternal prognosis, severe lethal congenital malformations).
Acknowledgements
We would like to thank everyone who has contributed to this study.
Ethical approval and consent to participate
The Ethics Committee for Biomedical Research, University Mohammed V – Rabat, Faculty of Medicine and Pharmacy – Rabat, approved the study, n°: 20/16. The oral consent to participate was obtained from the parents.
Funding
Not applicable
Competing interests
The authors declare that they have no competing interests.
Authors ‘contributions
ATI was responsible for the study concept and design. KF coordinated the study and interviewed the parents. KF and EB analyzed the data. KF led the drafting of this manuscript, with input from all authors. ATI and MHA supervised analysis of the data and contributed to the writing. ATI, MHA, EB, and AMA contributed to the revision and final approval of the manuscript.
All the authors have approved the present version of the manuscript.
References:
-
Marcorelles P, Laquerriere A. Neuropathology of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C (1):109–119. PMID: 20104606 http://doi : 10.1002 / ajmg.c.30249.
-
DeMyer W, Zeman W. Alobar holoprosencephaly (arhinencephaly) with median cleft lip and palate: clinical, electroencephalographic and nosologic considerations. Confiner Neurol. 1963; 23:1–36. PMID: 14026941. http://doi : 10.1159 / 000104278
-
DeMyer W, Zeman W, Palmer CG. Familial alobar holoprosencephaly (arhinencephaly) with median cleft lip and palate: report of a patient with 46 chromosomes. Neurology. 1963; 13:913–918. PMID: 14079950. http://doi: 10.1212 / wnl.13.11.913
-
DeMyer W, Zeman W, Palmer CG. The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly). Pediat. 1964; 34:256–263. PMID: 14211086
-
Mercier Sandra, Dubourg Christèle, [...], and Odent Sylvie. New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases. J Med Genet. 2011 Nov; 48(11): 752-760. PMID: 21940735 http://doi : 10.1136 / jmedgenet-2011-100339.
-
Winter Thomas C, Kennedy Anne M, Woodward Paula J. Holoprosencephaly: A Survey of the Entity. Embry Fetal Imaging.Radiogr. 2015;35(1):275–290. PMID: 25590404 http://doi : 10.1148 / rg.351140040.
-
Roessler E, Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010; 154C(1):52-61. PMID: 20104595 https://doi.org/10.1002/ajmg.c.30236
-
Hong Mingi and Krauss Robert S. Modeling the complex etiology of holoprosencephaly in mice. Am J Med Genet C Semin Med Genet. 2018 June; 178(2): 140–150. PMID: 29749693 http://doi:10.1002/ajmg.c.31611
-
Hong M, Krauss RS. Ethanol itself is a holoprosencephaly-inducing teratogen. PLoSOne. 2017; 12(4): e0176440.PMID: 28441416 https://doi.org/10.1371/journal.pone.0176440
-
Vaz SS, Chodirker B, Prasad C, Seabrook JA, Chudley AE, Prasad AN. Risk factors for nonsyndromic holoprosencephaly: a Manitoba case-control study. Am J Med Genet. 2012;158A(4):751–758.PMID: 22419615 http:// doi: 10.1002 / ajmg.a.35240
-
LekshmiARV, Raju G, Chandrakumari K.Foetal Anomalies: Correlative Study of Sonography and Autopsy.Int J Cur Res Rev. 2021;13(2):143-147 http://dx.doi.org/10.31782/IJCRR.2021.13218
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





