IJCRR - 8(7), April, 2016
Pages: 01-03
Date of Publication: 12-Apr-2016
Print Article
Download XML Download PDF
BARDET BEIDL SYNDROME - A RARE PLEIOTROPIC DISORDER
Author: Sonia Jaiswal, Navbir Pasricha, Prashant Bhargava
Category: Healthcare
Abstract:Background: Bardet\?Biedl Syndrome is a disorder with a pleiotropic gene action on multiple phenotypic traits and thus a wide range of clinical variability is seen within and between families. Bardet Beidl Syndrome is characterized by rod cone dystrophy, truncal obesity, postaxial polydactyly, cognitive impairment, male hypo-gonadotrophic hypogonadism, complex female genitourinary malformations and renal abnormalities. Case Presentation: A one day old male child came to the Outpatient department of a private clinic. The case presented with polydactyly, absence of hard palate due to failure of fusion of bilateral premaxillary segments and macular grade corneal opacity as examined by an ophthalmologist. Conclusion: The diagnosis of Bardet Beidl syndrome (BBS) is established by clinical findings. According to studies by Beales et al in 1999 and 2001, diagnosis is confirmed with the presence of three primary features and two secondary features. An interpretation of the molecular pathogenesis with a thorough research into therapeutics may bring about new treatment options for the organ specific disorders of Bardet Beidl Syndrome.
Keywords: Bardet beidl syndrome, Polydactyly, Syndactyly, Hypogonadism
Full Text:
INTRODUCTION Bardet Beidl syndrome is a multisystem autosomal recessive disorder. The syndrome is named after Georges Bardet and Arthur Beidl.(1) It is synonymous with Lawrence Moon syndrome but it is a matter of debate as some workers consider it as a separate entity while some recent research suggests that the two conditions may not be distinct .(2) Bardet-Biedl syndrome is a multisystemic genetic disorder characterized by postaxial polydactyly, progressive retinal dystrophy, obesity, hypogonadism, learning difficulty, and renal dysfunction. Other manifestations include diabetes mellitus, neurological impairments (mainly ataxia), heart disease, oro- dental malformations. Due to the late onset of symptoms, the diagnosis of BBS is usually made during childhood.
For example, obesity appears around age 2–3 years, and retinal degeneration becomes clinically apparent only at age 8 years.(3,4) The only features that may be present at birth are polydactyly, kidney anomaly, hepatic fibrosis, and genital or heart malformations. Bardet-Biedl syndrome (BBS) is a rare, genetic multisystem disorder; a ciliopathy secondary to the basal body dysfunction.(5,6) Mutations in 14 genes are known to be associated with BBS ( Bardet Beidl Syndrome): BBS1, BBS2, ARL6/ BBS3, BBS4, BBS5, MKKS/BBS6, BBS7, TTC8/ BBS8, B1/ BBS9, BBS10, TRIM32/BBS11, BBS12, MKS1/ BBS13, and CEP290/BBS14 .( 7)
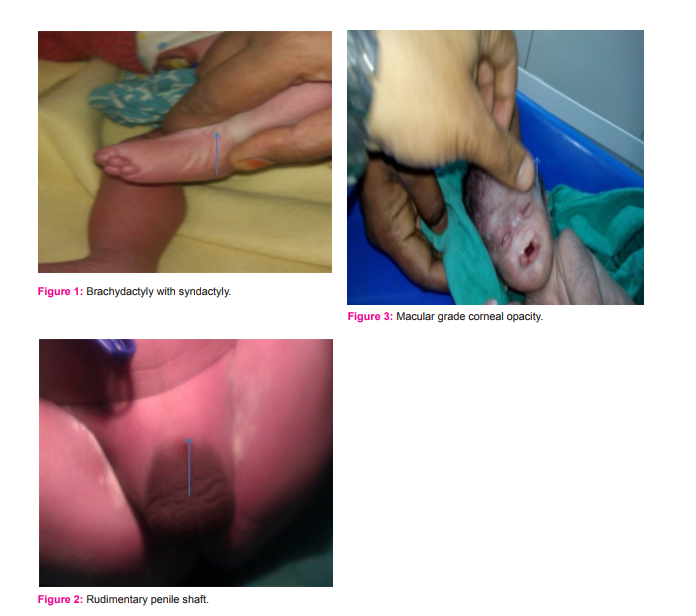
CASE PRESENTATION A one day old male baby with a normal birth weight of 2.5 kg came to a private clinic (Sai clinic). The father complained about the deteriorating condition of the patient due to fever and breathlessness. On careful examination by a Neonatologist the width of the head region was more as compared with the length, the back of head was flat rather than curved and the tips of ears were protruded. The patient had a flattened nasal bridge, there was presence of a bilateral cleft lip and palate and an anomaly was seen in the lower limb with hexadactyly in the left foot and brachydactyly with syndactyly in the right foot (fig 1). Hypogonadism was also present in the patient (rudimentary penile shaft was seen fig 2).
The Ophthalmologist observed a semi dense opacity in the cornea with the rest of the cornea hazyand hence diagnosed the condition as a segmental macular grade corneal opacity(fig 3). On the basis of the clinical findings a diagnosis of BardetBeidl syndrome was made. Beales et al (1999) and Beales et al (2001) have suggested that the presence of three to four primary features and two secondary features is a diagnostic feature of Bardet-Beidl Syndrome.(3,4)
DISCUSSION Bardet and Biedl first described BBS in the early 1920s. For many years it was considered as Laurence–Moon–Bardet– Biedl syndrome (MIM 245800), but now Lawrence Moon and Bardet Beidl syndromes are recognized as separate syndromes. Bardet-Biedl syndrome affects males and females in equal numbers. The prevalence is estimated to be 1 in 100,000 in the non-related (non-consanguineous) populations of Northern Europe and America. In Sweden, the prevalence is estimated to be 1 in 160,000. The disorder occurs with greater frequency in the Bedouin population of Kuwait (1 in 13,500) and in certain populations of Newfoundland (1 in 17,500).
The estimated incidence of this rare syndrome is 1/ 125 000– 160 005. (8) In almost all reported cases, digital abnormalities are invariably present. Kwitek-Black et al reported a large inbred Bedouin family from the Negev region of Israel with all nine affected subjects having polydactyly.(9) Green et al examined 32 patients with BBS and found that all cases (32) had syndactyly, whereas brachydactyly, polydactyly was present in 18 of 31 patients.(10) These findings are similar to our case with hexadactyly in the left foot and brachydactyly with syndactyly in the left foot.
Carmi et al found that there was a phenotypic variability in the number and severity of clinical manifestations in BBS(BardetBeidl Syndrome), particularly in the pattern of polydactyly thus reflecting the differing genotypes underlying the disorder.(11)Somwanshi reported four cases (three males, one female) with polydactyly, hypogonadism, retinitis pigmentosa, obesity, and mental retardation,(12) similarly the male infant in our case presented with hypogonadism. The most common defective gene associated with BardetBiedl syndrome is the BBS1 gene located on the long arm (q) of chromosome 11 (11q13). The BBS1 gene accounts for approximately 30 percent of cases. The genes associated for Bardet-Biedl syndrome contain instructions for encoding proteins.
Mutations of these genes results in altered function of these proteins. Investigators have determined that most of the Bardet-Biedl proteins are associated with cilia which are hair-like structures that cover most type of cells in the body, and structures such as the basal body (which help in anchoring the cilia to a cell) or flagella. Recent findings in genetic research have suggested that a large number of genetic disorders including both syndromes and diseases have perhaps no relation phenotypically but may be related genotypically.
BBS is one such syndrome that has now been identified to be caused by defects in the cellular ciliary structure thus making it a ciliopathy. Other known ciliopathies include primary ciliary dyskinesia, polycystic kidney and liver disease, nephronophthisis, Alstrom syndrome, Meckel– Gruber syndrome and some forms of retinal degeneration.(13)
CONCLUSION The diagnosis of BBS in this case was made on the basis of clinical features. The mechanism of the clinical and genetic diversity in BBS patients is not yet known. A careful clinical evaluation of the molecular pathogenesis of BBS will help in a better understanding of this disorder.
References:
1. Synd/3745 at who named it?
2. Moore S, Green J, Fan Y; et al. (2005). “Clinical and genetic epidemiology of Bardet–Biedl syndrome in Newfoundland: a 22- year prospective, population-based, cohort study”. Am. J. Med. Genet ARRAY 132 (4): 352– 60. doi:10.1002/ajmg.a.30406. PMC 3295827. PMID 15637713.
3. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA (1999) New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet36:437– 446
4. Beales PL, Katsanis N, Lewis RA, Ansley SJ, Elcioglu N, Raza J, Woods MO, Green JS, Parfrey PS, Davidson WS, Lupski JR (2001) Genetic and mutational analyses of a large multiethnic Bardet-Biedl cohort reveal a minor involvement of BBS6 and delineate the critical intervals of other loci. Am J Hum Genet 68:606–616
5. Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, Mah AK, Johnsen RC, Cavender JC, Lewis RA, Leroux MR, Beales PL, Katsanis N: Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 2003, 425:628-633.
6. Adams M, Smith UM, Logan CV, Johnson CA: Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet 2008, 45:257-267.
7. Waters AM, Beales PL: Bardet-Biedl Syndrome.[http://www. ncbi.nlm.nih. gov/books/NBK1363/].
8. Klein D, Ammann F. The syndrome of Laurence–Moon–Bardet– Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. Neurol Sci 1969; 9: 479–513.
9. Kwitek-Black AE, Carmi R, Duyk GM, Buetow KH, Elbedour K, Parvari R, Yandava CN, Stone EM, Sheffield VC. Linkage of Bardet– Biedl syndrome to chromosome 16q and evidence for non-allelic genetic heterogeneity. Nat Genet 1993; 5: 392–6
10. Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O’Leary E, Pryse-Phillips W. The cardinal manifestations of Bardet–Biedl syndrome, a form of Laurence–Moon– Biedl syndrome. New Eng J Med 1989; 321: 1002–9.
11. Carmi R, Elbedour K, Stone EM, Sheffield VC. Phenotypic differences among patients with Bardet–Biedl syndrome linked to three different chromosome loci. Am J Med Genet 1995; 59: 199– 203
12. Somwanshi PR, Nikam SH, Patni PD: Laurence Moon Biedl Bardet syndrome. J Assoc Physician India 1988, 36:333-335.
13. Badano JL, Mitsuma N, Beales PL, Katsanis N (2006). “The ciliopathies: an emerging class of human genetic disorders”. Annu Rev Genomics Hum Genet 7: 125–48. doi:10.1146/annurev.genom.7.080505.115610. PMID 16722803.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





