IJCRR - 8(8), April, 2016
Pages: 08-14
Date of Publication: 20-Apr-2016
Print Article
Download XML Download PDF
BIOAVAILABILITY AND BIOEQUIVALENCE EVALUATION OF SOME GENERIC PRODUCTS OF ARTEMETHER-LUMEFANTRINE DOUBLE STRENGTH TABLETS MARKETED IN NIGERIA
Author: Awofisayo Sunday O., Okhamafe Augustine O., Arhewoh Matthew I.
Category: Healthcare
Abstract:The study was aimed at evaluating the bioavailability and bioequivalence of generic products of artemether-lumefantrine (AL) antimalarial double strength oral tablet formulation. A non-randomized open label single dose study in eighteen healthy African male subjects was designed. The volunteers were administered one tablet of a product with a fatty meal and 0.5 L of water, after overnight fast. Venous blood sampling was taken at 0, 1, 2, 4, 6 and 8 h post-dose and plasma samples analyzed for artemether and lumefantrine exposure simultaneously using a validated high performance liquid chromatographic system with Chromosil C18 column, flow rate and UV detection at 1.0 mL/min and 216 nm, respectively. Acetonitrile: potassium dihydrogen phosphate (70: 30%, v/v) and nevirapine were employed as mobile phase and internal standard, respectively. The primary endpoints were area under the plasma concentration time curve (AUC) from zero to 8 h and maximum plasma concentration (Cmax). The 90% confidence interval for the ratio of the geometric means of AUC0-8 was compared with the established bioequivalence limit. All enrolled subjects with mean age 28.5 \? 4.5 years completed the study. The Cmax for artemether and lumefantrine for the products ranged from 0.225 - 0.558 µg/mL and 0.319 \? 0.517 µg/mL, respectively. Drug products AL1 and AL2 met the bioequivalence criteria. The other products failed the pharmacopoeia test specification for chemical content with respect to either or both API and their pharmacokinetic profiles varied with statistical significant differences (P>0.05). Only two of the generic products studied were bioequivalent and could be switched.
Keywords: Bioavailability, Bioequivalence, Pharmacokinetics, Artemether, Lumefantrine
Full Text:
INTRODUCTION Oral administration of drugs is convenient and this route forms the largest means of drug administration (Martinez and Amidon, 2001). To produce a clinical response, drugs are required to achieve an effective concentration at the site of action and this maintained for an adequate length of time (Abdou, 1989). Orally administered drugs required for systemic agents, involves the transfer of the drugs from the gut to the systemic circulation. Bioavailability describes the fraction of the drug dose that reaches the systemic circulation unchanged. As many drugs are made and marketed by more than one pharmaceutical company, the application of biopharmaceutics gives substantial evidence on the effect of the method of manufacture (i.e., procedures and materials) on bioavailability of the drug (Chow and Liu, 1992).
The plethora of drug products containing the same active ingredients and possibly different excipients may require that all concerned (i.e., physician, pharmacists and end-users) select products that produce equivalent therapeutic effects. Most bioavailability studies, either for new or generic product, require qualitative tests to assess the performance of orally administered formulations (Pabst and Jaeger, 1990]. Some useful guidelines and methods have been developed by the U.S. Food and Drug Administration for determining drug bioavailability (Ribeiro et al., 2000). Bioavailability studies are therefore important in defining the safety and efficacy of drug products in terms of its effect on drug pharmacokinetics while bioequivalence studies compares the bioavailability of a drug from various drug products (USP, 2001)
In cases where there is approval of multisource marketing of any particular product, generic substitution and dispensing of different brands or unbranded products in place of the prescribed products may prevail without sufficient evidence of pharmaceutical equivalence or assurance of therapeutic performance (Committee for Proprietary Medicinal Products, 1992). Artemether-lumefantrine is the most widely employed fixeddose artemisinin combination therapy (ACT) employed for the treatment of uncomplicated malaria (FDA-CDER, 2002; WHO, 2013). In Nigeria, the National Guidelines on Malaria Treatment, Prevention and Control recommends, and in principles, supports the multisource availability of AL products (Dondorp et al., 2005). The national drug regulatory agency in Nigeria has similarly approved for marketing, in recent times, DS formulations but speculations as to the bioavailability and bioequivalence indices of the several marketed products has been raised. In previous studies on bioavailability of AL generic tablet formulation conducted in Tanzania, it was concluded that the bioequivalence was not demonstrated due to non-compliance with FDA 90% confidence interval criteria (Federal Republic of Nigeria, 2005). This study was aimed at assessing the bioavailability and bioequivalence of some AL DS tablet formulation marketed in Nigeria.
MATERIALS AND METHODS Chemicals HPLC grade acetonitrile and potassium dihydrogen phosphate (Sigma-Aldrich, Germany) were used for the LC analysis. Nevirapine powder, artemether and lumefantrine reference standards were donated by Central Medical Laboratory of University of Lagos (CMUL). Deionized water was used throughout and obtained from a Milli-Q system (Millipore, MA, USA). The generic products of artemether-lumefantrine were purchased in a licensed pharmacy outfit in Uyo, Nigeria. Details of the generic products are presented in Table 1.
All other reagents were of analytical grade. Chromatographic system Simultaneous determination of artemether and lumefantrine was done using HPLC (HPLC Peak 7000 System (Rheodyne manual sample injector, analytical Chromosil column C18, 250x4.6 mm column, Germany) with wavelength of UV detection at 216 nm and nevirapine as internal standard. The mobile phase was a mixture of acetonitrile and 25 mM potassium dihydrogen phosphate (70:30%, v/v). The total run time was 8 min. The method of analysis was developed by the CMUL and modified from an earlier reported method by Cesar and co-workers (International Conference on Harmonization, 1995). Study design The study had an open label randomized one-period and single-dose design. Eighteen healthy volunteers were randomly assigned to one of six AL DS generic products.
The study was conducted in accordance with the Declaration of Helsinki 2013 and Good Clinical Practices Guidelines set up by the International Conference on Harmonization and local applicable laws and regulations (Minzi et al., 2013). Subject Recruitment and Management Volunteers were eligible for the study if they had a negative result for malaria parasite test (using microscopic detection of parasites in blood smear after Giemsa stain) and had not taken any antimalarial in the past 2 months from the commencement of the study. Other eligibility criteria included Haemoglobin level >9.0 g/dL (5.5 mmol/L, serum bilirubin ≤ 2 mg/dL (34 µmol/L) and adequate renal function defined as serum creatinine clearance ≥ 60 mL/min. Volunteers were excluded if they had gross ascites or an evidence of active peptic ulcer disease or other documented gastrointestinal (GI) diseases that might influence GI motility (Cesar et al., 2008).
They were certified healthy by a physician based on their medical history, clinical examination, haematological and biochemical screening. The study protocol was approved by the Institutional Health Research Committee of University of Uyo Teaching Hospital (approval certificate number UUTH/AD/S/96/VOL.XVI/67). All volunteers gave written informed consent. The study was done according to the Guidelines of Helsinki Declaration, 2013 (World Medical Association, 2013).
Drug products and administration Volunteers received a single dose of the drug after an overnight fast, administered with 500 mL of water within 15 min of finishing breakfast (a standard measure of oily rice). Food and water intake was restricted for 4 h after ingestion of AL tablet. Blood sample collection Blood samples (5 mL each), taken from an indwelling intravenous cannula placed in the arm were collected in heparinized tubes at 0, 1, 2, 4, 6 and 8 h post dose. The samples were centrifuged at 3000 rev/min for 5 min and the clear supernatant transferred to a polypropylene tube and immediately stored at -60o C until analysis.
Pharmacokinetic analysis The maximum drug concentration (Cmax), time to achieve the maximum concentration (Tmax) and area under the curve (AUC0-8) were determined by imputing the experimental data for each subject in each group into GraphPad Prism pharmacokinetic software (Prism for Windows version 6.05, USA). The pharmacokinetic parameters (AUC0-8, Cmax, Tmax) for the AL tablet formulations were compared with that of the established product. Statistical analysis Minitab for Windows Statistical Package (Minitab Inc., USA), was used for statistical analysis. The statistical analysis employed single factor one way analysis of variance (ANOVA).
The parameters C max and AUC (0-8) were computed for individuals and the mean for each group was tested. The null hypothesis of no difference amongst the means of parameters for the generic products was tested at a 5% level of significance. Classical 90% confidence Intervals (CI) were calculated for the parameters based on the currently accepted criteria for assessing bioequivalence (Cesar et al., 2008). To test for differences amongst the generic products, post hoc multiple comparison was performed using Duncan Multiple Range (DMR) and Student Neuman Keul (SNK).
Pharmacokinetic analysis The maximum drug concentration (Cmax), time to achieve the maximum concentration (Tmax) and area under the curve (AUC0-8) were determined by imputing the experimental data for each subject in each group into GraphPad Prism pharmacokinetic software (Prism for Windows version 6.05, USA). The pharmacokinetic parameters (AUC0-8, Cmax, Tmax) for the AL tablet formulations were compared with that of the established product. Statistical analysis Minitab for Windows Statistical Package (Minitab Inc., USA), was used for statistical analysis.
The statistical analysis employed single factor one way analysis of variance (ANOVA). The parameters C max and AUC (0-8) were computed for individuals and the mean for each group was tested. The null hypothesis of no difference amongst the means of parameters for the generic products was tested at a 5% level of significance. Classical 90% confidence Intervals (CI) were calculated for the parameters based on the currently accepted criteria for assessing bioequivalence (Cesar et al., 2008). To test for differences amongst the generic products, post hoc multiple comparison was performed using Duncan Multiple Range (DMR) and Student Neuman Keul (SNK).
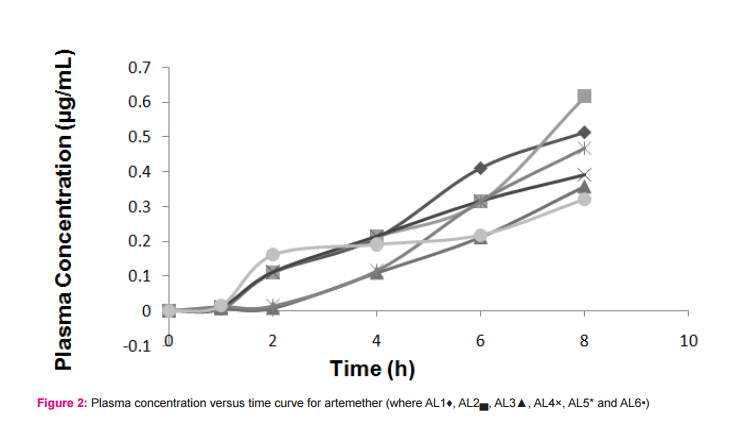
RESULTS AND DISCUSSION Malaria is a common parasitic disease known across the globe with all attention geared at combating the menace. The multisource proliferation of the generic products of antimalarial agents will therefore require establishing their comparable drug release and bioequivalence. Analysis of drug samples The result of the chemical content of the drug samples is presented in Table 2. Samples AL1 and AL2 had satisfactory drug contents with respect to the active ingredients. Samples AL3, AL4, AL5 and AL6 failed the Pharmacopoeia specification for chemical content with values less than 90% content for lumefantrine (USP, 1990). Volunteers and treatment Eighteen healthy male volunteers were enrolled for the study. The median age was 28 (and range, 26-34) years, mean body weight and mean body mass index (BMI) of 76.5 Kg and 27.5 Kg/m2 , respectively. Overall, the drug was well-tolerated. Figures 2 and 3 presented the plasma concentration versus time curves for the drug products with respect to artemether and lumefantrine, respectively. The resulting pharmacokinetic parameters for the various sampled drugs are presented in Table 3. The study was conducted in accordance with the Declaration of Helsinki 2002 (Henderson et al., 1992; International Conference on Harmonization, 1995), Good Clinical Practices Guidelines set up by the International Conference on Harmonization and local applicable laws and regulations. The study here provides a simultaneous determination of the active pharmaceutical ingredients (i.e., artemether and lumefantrine) components of the drug. Previous work by Souppart et al., evaluated the plasma levels of artemether using HPLC-MS (Souppart et al., 2002) and another work involving LC-MS/MS method for the determination of lumefantrine and its metabolite desbutyl lumefantrine in plasma from patients infected with Plasmodium falciparum malaria (Sethi et al., 2011).
This present work evaluates the selected products based on the simultaneous determination of the unchanged drugs (i.e., artemether and lumefantrine components) in the dosage form and in plasma. Following up on the bioequivalence of artemether-lumefantrine drug products through artemether exposure has presented some difficulties because on the instability of this drug component. The use of high performance liquid chromatographic system involving nevirapine as internal standard, as employed in this study, was able to resolve the variability issues associated with artemether instabilities/analytical handling complexities. Previous work employed halofantrine for the single drug component analysis of lumefantrine (Huang et al., 2012). The wavelength of UV detection in the work by Huang et al. was 335 nm while in this study involving nevirapine as internal standard, UV detection was at 216.
Nevirapine gave signals with shorter resolution time distinct from those of the analytes of interest. Periodically interfering peaks have been reported with the analytes when halofantrine is used as internal standard (Huang et al., 2012). As at the time of this study, the patent right owners of AL markets only the 20/120 tablet formulation hence AL1 being the established and most widely prescribed generic, was referred to as the reference product. AL1 also gave a satisfactory chemical content that met the pharmacopoeia standard. The comparative bioavailability of the AL DS generics revealed that only product AL2 met the bioequivalence criteria by possessYing a relative bioavailability of 0.85. The calibration curve for artemether and lumefantrine determination in plasma gave a correlation coefficient of 0.98 and 0.94 respectively.
The values were considered satisfactory for drug absorption kinetics involving plasma samples, so much more with the use of an internal standard in the analysis. The chemical content of the AL generic products Table 2 revealed that only products AL1 and AL2 were pharmaceuti- cal equivalents with respect to both component drugs. The other products failed the chemical content test with respect to lumefantrine whereas AL6 failed with respect to both drug components. Bioequivalence was therefore not considered for these products regardless of their relative bioavailability values. Several antimalarial products that are artemisinin-based have been reported with lower than required concentration of active ingredients (Kaur et al., 2008; Nayyar et al., 2012). It is however required that products considered for bioequivalence meet pharmacopoeia standards before being evaluated for bioequivalence with reference product (Ribiero et al, 2000). Products AL3, AL4, AL5 and AL6 are thus not meet for bioequivalence considerations.
CONCLUSION Many antimalarial agents in use have been reported to fall below the regulatory standards due to deliberate counterfeiting, poor quality control during manufacture or decomposition due to improper handling or storage. The wide difference in the chemical content and gross relative bioavailability of the selected products serve as a pointer to the causes of the nagging issue of drug resistance and failure in malaria treatment.
ACKNOWLEDGEMENT The authors are grateful to Mr. P. Ojobor for his technical assistance. AUTHOR’S STATEMENT The authors have no conflict of interest to disclose.

References:
1. Abdou H.M. 1989. Dissolution, bioavailability and Bioequivalence. Easton: MACK Publishing Company.
2. Cesar I.C., Nogueira F.H.A. and Pianetti G.A. 2008. Simultaneous determination of artemether-lumefantrine in fixed dose combination tablets by HPLC with UV detection. J Pharm and Biomed Anal 48: 951-954.
3. Chow S.C., Liu J.P. 1992. Design and Analysis of Bioavailability and Bioequivalence Studies. New York: Marcel and Dekker, Inc.
4. Committee for Proprietary Medicinal Products. 1992. Notes for Guidance Investigation of Bioavailability and Bioequivalence (III/54/89/EN), CPMP, Brussels.
5. Dondorp A., Nosten F., Stepniewska K., Day N. and White N. 2005. South East Asian Quinine Artesunate Malaria Trial (SEAQUAMAT) group. Artesunate versus quinine for treatment of severe falciparum malaria: a randomized trial. Lancet. 366:717- 725.
6. FDA US Food and Drug Administration, CDER (Center for Drug Evaluation and Research). 2002. Guidance for Industry: Food Effect bioavailability and fed bioequivalence studies.
7. Federal Republic of Nigeria. 2005. National Antimalarial Treatment Policy. Federal Ministry of Health, National Malaria and Vector Control Division, Abuja, Nigeria, Pp 1-35.
8. Guidelines on the design of a single-dose in-vivo bioavailability study, Bioavailability and Bioequivalence Requirements, 21 CFR. 1991. 320:26.
9. Henderson J.D., Dighe S.V., Williams P.L. 1992. Subject selection and management in bioequivalence studies. Clinical Research and Regulatory Affairs 9: 71-87.
10. Huang L., Li X., Marzan F., Lizak P.S., Aweeka T.A. 2012. Determination of lumefantrine in small-volume human plasma by LC-MS/MS using deuteriated lumefantrine to overcome matrix effect and ionization saturation. Bio-analysis 4: 157-166.
11. International Conference on Harmonization. 1995. Structure and Content of Clinical Study Report; E3.
12. Kaur H., Goodman C., Thompson E., Thompson K.A., Masanya I. et al. 2008. A nationwide Survey of the Quality of Antimalarials in Retail Outlets in Tanzania. PLoS ONE 3: 1-7.
13. Martinez M.N. and Amidon G.L. 2001. A Mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol 42: 620 – 43.
14. Minzi O.M.S., Maraelle I.A., Shekalaghe S., Juma O., Ngaimisi E. et al. 2013. Comparison of bioavailability between the most available generic tablet formulation containing artemether and lumefantrine on the Tanzanian market and the innovator’s product. Mal J 12: 174-186.
15. Nayyar G., Bremen J.G., Newton P.N. and Herrington J. 2012. Poor-quality antimalarial drugs in Southeast Asia and Sub-Saharan Africa. Lancet Infect Dis. 12: 488-496.
16. Pabst G., Jaeger H. 1990. Review of methods and criteria for the evaluation of bioequivalence studies. Eur J Cancer 38:5-8.
17. Ribeiro W., Zappi E.A., Moraes M.E., Bezerra F.A., Lerner F.E. et al., 2000. Comparative bioavailability of two fluconazole capsule formulations in healthy volunteers. Arzneimittelforschung 50:1028-1032.
18. Sethi P., Dua V.K., Jain R. 2011. A LC-MS/MS method for the determination of lumefantrine and its metabolite desbutyl lumefantrine in plasma from patients infected with Plasmodium falciparum malaria. J Liq Chromatr and Related Tech 34:25-35.
19. Souppart C., Gauducheau N., Samdrenam N., Richard F. 2002. Development and validation of a high performance liquid chromatography- mass spectrometry assay for the determination of artemether and its metabolite dihydroartemisinin in human plasma. J Chromatography B 774: 195-203.
20. USP XXII/NFXVII. 1990. United States Pharmacopoeia Convention, Inc., Rockville, Maryland. Pp1578-1579.
21. USP. 2001 In-vitro and In-vivo evaluation of dosage forms . In United States Pharmacopeias and National formulary USP 26 – NF 21. United States Pharmacopeia Convention, Inc.: Rockville, MD.
22. World Health Organization. 2013. World Malaria Report; Geneva, Switzerland; http:// www.WHO. Int/malaria/world_malaria_report_2013/en/ (Accessed on June16, 2014).
23. World Medical Association. 2013. Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. JAMA 3100 (2): 2191-94.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





