IJCRR - 10(10), May, 2018
Pages: 19-26
Date of Publication: 30-May-2018
Print Article
Download XML Download PDF
Demographic, Clinical and Etiological Profile of Young Dystonia Less Than Forty Years of Age \? A Hospital Based Study
Author: S. Bashir Ahmad, Bashir Z., Tak S., Goyal V., Behari M.
Category: Healthcare
Abstract:Objective: To study the demographic, clinical and etiological profile of dystonia in patients less than forty years of age.
Material and Methods: In this study two hundred and nineteen patients with different dystonia were included. Data on demographic profile, clinical and etiological profile was recorded and statistical analysis was done by using descriptive analysis (frequency distribution).
Results: Out of two hundred and nineteen patients, one hundred thirteen (51.7%) were < 26 years of age of onset and one hundred six (48.4%) had >26 years of age of onset with mean age of onset 22.3 years, one hundred seventy six were males and forty four females. Mean duration of illness was 4.4 years. Frequency of different dystonia was focal dystonia 57.72%, multifocal 1.36%, segmental 10%, generalized 29.09%, hemidystonia 3.63% and paroxysmal kinesigenic dystonia 2.72%. Among focal dystoniaWriter's cramp was most common (63%) followed by cervical dystonia (17.32%), and blepharospasm (7%). Among generalized dystonia Wilson's disease was most common etiological factor in 31.25% cases. Primary generalized dystonia was seen in 17.2% cases. MRI was abnormal in all eight cases of hemidystonia. SPECT brain was abnormal in 1 out of four cases of paroxysmal kinesigenic dystonia. DYT1 was positive in four out of eleven patients of primary generalized dystonia.
Conclusion: Present study showed that focal dystonia were more common even in younger age group with Writers cramp most common among all dystonia followed by cervical dystonia. In hemidystonia structural lesion should always be ruled out.
Keywords: Dystonia, Paroxysmal kinesigenic dystonia, Writer’s cramp
Full Text:
INTRODUCTION
There have been only few studies on demographic, clinical and etiological profile of young dystonia in less than forty years of age group from India in English literature. Dystonia is defined as a syndrome of sustained muscle contraction, frequently causing twisting and repetitive movements or abnormal postures. It can be jerky, rhythmic or tremulous1.
In 1911, Oppenheim introduced term dystonia and used two different names for this syndrome, dysbasialordotica progressive and dystonia muscularamdeformens1. Flater and Sterlig emphasized its organicity and likely to be hereditary2. Since dystonia can have so many etiologies, the true prevalence is unknown.
Epidemiological survey of dystonia has shown that it is the third most prevalent movement disorder after Parkinson's disease and essential tremor3. Marsden estimated that one fourth of patients have a secondary dystonia and rest are primary or idiopathic4.
Due to clinical and etiological heterogeneity of dystonia and in recent years their classification has been reviewed leading to an etiological approach where they are classified on the basis of age of onset, by distribution, temporal pattern, associated features and cause5. Clues to secondary dystonia are, associated features, begin suddenly, occur at rest, environmental causes, cranial onset in young and lower limb onset in adults, drug or toxin exposure, past history of encephalitis, trauma, cerebral palsy, meningitis and abnormal cranial imaging6.
Primary dystonia is a complex network disorder arising from the dysfunction of one or more parts of the brain including the basal ganglia, thalami, cortex, cerebellum, pons and dentate nuclei. Distorted proprioceptive propricephic feedback from muscle spindle to central process of structures7.
This was confirmed by Magayar-Lehman et al on PET studies8. Fahn et al found that the yield of investigate in childhood, adolescent and adult onset dystonia was 41%, 32% and 13% respectively9. Majority of hemidystonia has secondary cause10. Clinical presentation, demographic and etiological profile is different in younger age group patients as compared to adults11.
OBJECTIVES
Objectives of the present study was to study the demographic, etiological and clinical profile in different dystonia and in patients of <40 years of age of onset.
MATERIAL AND METHODS
This prospective study was conducted in the Departments of Neurology, Superspeciality Hospital of Government Medical College Srinagar and AIIMS New Delhi and includes patients from outpatient and inpatients departments. Enrollment of patients was done from May 2003 to January 2014. Patients of less than forty years of age with diagnosis of dystonia were included in the study. Patients enrolled in the study were subjected to detailed clinical history and general physical examination to look for the evidence for KF ring, telangiectasias and signs of chronic liver disease. Directed systemic examination was done to look for any evidence of organomegaly, contractures and abnormal postures. Detailed neurological examination was done in all patients to look for any associated neurological deficits.
Investigations included hemogram, liver function tests, kidney function tests, uric acid, peripheral blood film for acanthocytes (3 consecutive samples and more than 10% was taken significant), Wilson profile (24 hour urinary copper and serum ceruloplasmin) and MRI brain. Genetic analysis for DYT1 was done in patients with primary generalized dystonia and less than twenty six years of age. Vasculitic profile was done in selected cases. MRI could be done also hundred patients and were discussed with neuro-radiologist. Any abnormality on MRI was recorded. Interictaland ictal SPECT brain was done in six paroxysmal kinesigenic dyskinesia patients. Dystonia was graded as per the dystonia rating scale described by Fahn (Appendix A).12
All variables were entered into the standard proforma designed at the start of the study. Data was collected of all patients enrolled for the study and was subjected for final analysis.
Statistical Methods
Descriptive analysis (frequency distribution) was used for final data analysis and results were depicted as percentage.
RESULTS
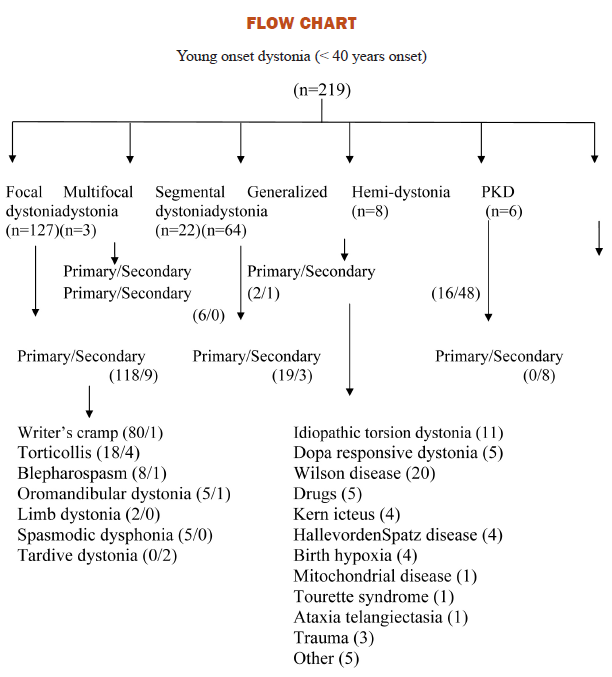
Total of six hundred cases of dystonia were registered out of which three hundred thirty (55%) were less than forty years of age and two hundred seventy (45%) were more than forty years of age. We could evaluate two hundred nineteen (66%) patients with different dystonia with age of onset less than 40 years. One hundred and fourteen (51.6%) had onset <26 years of age and the rest 48.7% had onset >26 years of age. Mean age at presentation was 22.3 years. One hundred seventy six (80%) patients were males and forty four (20%) were females. Mean duration of symptoms was 4.4 years (range 3-30 years). Onset in two hundred and fifteen (98.17%) was insidious and four (1.83%) had sudden onset. All these 4 patients had hemidystonia and were due to ischemic stroke, seven (3.2%)patients had diurnal variation and six of these had clinical suspicion of dopa responsive dystonia. One patient had primary generalized dystonia. One hundred five (42.7%) patients had only action/posture induced dystonia, three (1.4%) had at rest and hundred twelve (50.9%) had both at rest and during action (Table 1).
Ten (4.5%) patients had family history in 10 and 20 relatives (eight had dystonia in siblings, one in mother and one in father). Four patients (1.8%) with generalized dystonia had significant delayed milestones and among them three had onset before 26 years of age and one had after 26 years of age. Twenty five (11%) had significant past history in the form of stroke (twelve cases), trauma (six cases), chronic meningitis (three cases) and encephalitis (four cases). Ten patients (4.5%) had exposure to drugs like antiepileptics including (phenytoin) and 7 (3.9%) patients had exposure to neuroleptics including (risperidone, haloperidol and thioridazone). Six patients (2.7%) had history of orolingualself mutilation and only one patient showed evidence of acanthocytosis (Flow chart).
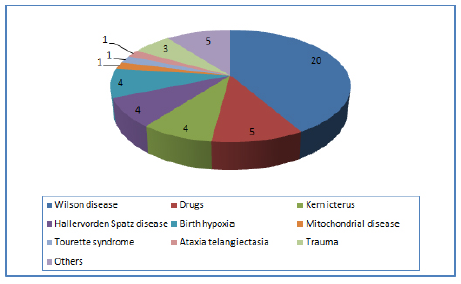
Ten patients (4.5%) had deranged liver function tests. Twenty five (11.4%) had abnormal copper studies. Out of these twenty five cases, twenty were diagnosed clinically as Wilson disease.
MRI was done in hundred patients (41.3%) and was abnormal in thirty seven patients showing abnormal signal changes in basal ganglia including striatum. Out of twenty patients of Wilson disease eighteen were having T2w signal changes in basal ganglia, thalami, dentate nucleus and cerebral white matter. In five patients with hemidystonia MRI showed infarcts in the basal ganglia region on the opposite side. Four patients with Hallervordenspatz disease had on MRI “eye of tiger sign” positive. Four patients had vascular malformation in the brain stem. Three patients with history of head injury had evidence of cortical scars on MRI brain. One patient with hypoxic brain insult had periventricular white matter abnormalities. One patient with history of tubercular meningitis had mild ventriculomegaly and T2w signal changes in bilateral basal ganglia.
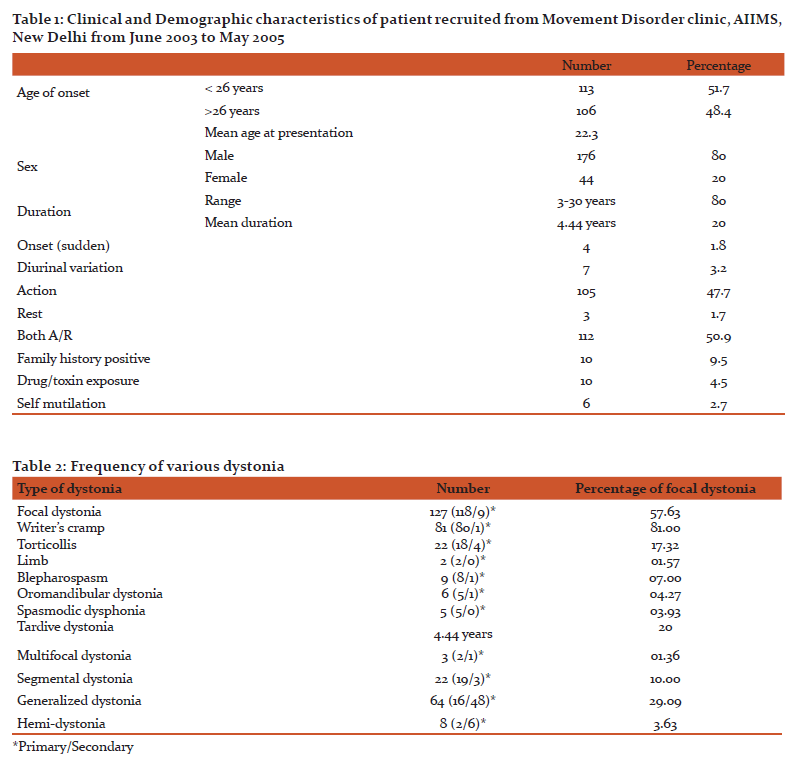
Among one hundred twenty seven cases of focal dystonia (57.99%), nine cases (7%) were secondary (Wilson disease = three, Trauma = two, Drugs = two, Vascular malformation = one, head injury = one). Of these writers cramp was seen in one, torticollis four, oromandibular dyskinesia two, tardive dystonia two.
Eight had hemidystonia and four were due to stroke on the opposite side, one had Wilson disease, two were post encephalitic and 1 was post head injury. Six patients (2.7%) had paroxysmal kinesgenic dyskinesia and one patients showed lesion in the contralateral frontal lobe on inter-ictal and ictal SPECT. Four patients (1.8%) had clinical and radiological evidence of Hallervordenspatz disease and all presented with generalized dystonia. Eleven patients (5%) had clinical suspension of idiopathic torsion dystonia and out of these four were DYT1 positive.
DISCUSSION
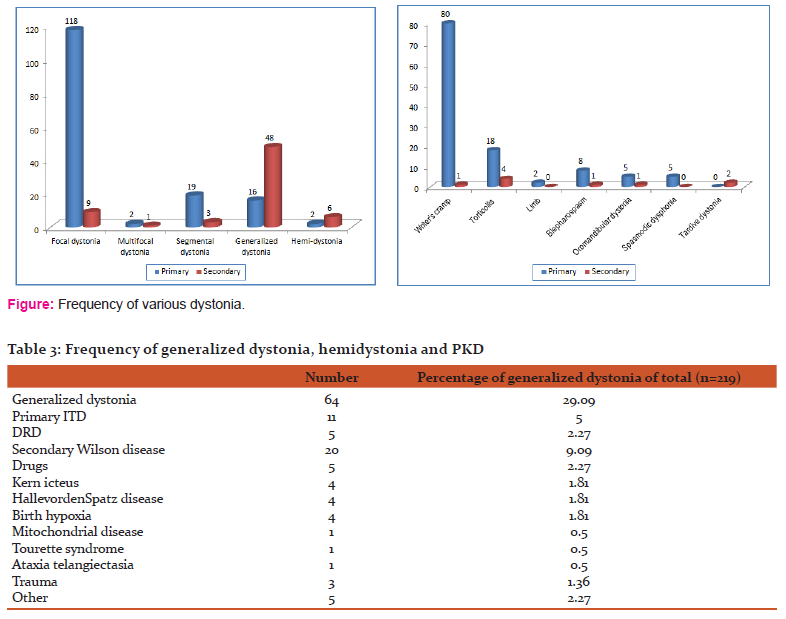
Present study was designed to see the demographic, clinical and etiological profile of dystonia in young age group (less than 40 years of age). This study is first of its kind in Indian population till date where all factor like demographic, clinical etiological and genetic profile has been studied in younger dystonia population less than forty years. Genetic analysis was done in patients who were less than twenty six years of age and who gave consent for the DYT1 gene estimation. Gene estimation at present is costly and most of our patient could not afford. So we stressed more on the demographic and clinical aspect of the dystonia. In present study focal dystonia were more common in<40 years age group (57.72%), followed by generalized dystonia in 29.9% cases,segmental 10%, hemodystonia 3.63%, paroxysmal kinesigenic dystonia (2.7%) and multifocal dystonia (1.36%) cases. Writers cramp was more common comprising 63% of all focal dystonia followed by torticollis 17% and blepharospasm in 7%. Primary dystonia was seen in 25% and secondary in 75%. Wilson disease was etiological factor in 41.7% of secondary generalized dystonia cases followed by drugs in 10.4%, Hallervorden Spatz disease in 8.4%, rest causes were less common. In 74.55% of focal dystonia and 25.45% generalized dystoniano cause could be found. Among one hundred thirteen patients with age of onset < 26 years, Fifty eight (50.87%) were focal dystonia, forty eight (42.10%) had generalized dystonia, four (3.5%) had hemidystonia and four (3.5%) segmental dytonia. Among one hundred six patients with age of onset > 26 years seventy five (70.75%) had focal dystonia, eighteen (16.48%) had generalized dystonia, seven (6.60%) had segmental dystonia, five (4.71%) had hemidystonia and one (0.94%) had multifocal dystonia.
Genetic analysis DYT1 estimation was possible in ten cases of 11 suspected idiopathic torsion dystonia and in four cases it was positive. Strong family history of dystonia was in ten cases.
According to an earlier study by Nutt JF et al13 cervical dystonia was the most common form of all the focal dystonia. This is contrary to our study where most common type of all focal dystonia was writer's cramp. The reason could be referral bias as patient with writer's cramp, are usually students and earning members of family. Disability due to writer's cramp, compels them to come for treatment at higher centers whereas patients with torticollis may not come for treatment. Also the population in the study reported included all groups of patients with focal dystonia whereas in our study we included patients < 40 years of age. Idiopathic or sporadic primary dystonia is the most common form of dystonia in most epidemiological or clinical population-based surveys. The multiple classification schemes for dystonia reflect clinical heterogeneity as well as the incomplete understanding of many of the underlying causes. When confronted with a new patient with dystonia, it is often helpful to consider first the descriptive feature of the condition before proceeding to identify genetic or other factors that may be causative (Bressman SB et al)14.
The most common cause of early onset generalized dystonia is DYT1 dystonia caused by a three base pair deletion (GAG deletion) in the TORIA gene. This mutation accounts for roughly 90% of early onset generalized dystonia in patients of Ashkenazi Jewish descent and approximately 40-60% in the non-Jewish population (Ozelius LJ et al)15. In our study, we could not perform genetic study in all patients with primary generalized dystonia due to high cost factor.
Chuang C et al16 evaluated the natural history in hemidystonia. One hundred ninety cases were identified and collected data on aetiology, age of onset, latency, lesion location and response to treatment. They found most common etiologies of hemidystonia were stroke, trauma and perinatural injury and mean age of onset was twenty years and mean latency from the insult to dystonia was 4.1 years and basal ganglia was the site of lesion in 48%. In present study with 8 cases of hemi-dystonia, six had stroke, one was post trauma and 1 post encephalitic.
Defazio G et al17 studied the risk factors for primary adult dystonia in two hundred and two cases (seventy three men and one hundred twenty nine women) with age range of 20-70 years. Focal dystonia was found in one hundred and fifty eight (78%) cases, forty four had cervical dystonia, twenty three with arm dystonia and one with leg dystonia, forty four with segmental and multifocal dystonia, thirty two cases with bleparospasm, twenty two with oromandibular, fifteen with cervical and five with laryngeal dystonia and one with leg dystonia. In thirteen cases of cervical dystonia had associated tremor. Six patients had dystonia at more than two sites. In one hundred twenty two patients cranial imaging was available which were normal. In the present study also there were more cases of focal dystonia, however writer’s cramp formed the majority.
Pettigrew L et al18 studied three hundred and nine patients of dystonia and observed twenty two with hemidystonia. All twenty two patients had acquired hemidystonia due to structural brain lesion, storage disease or degenerative neurological disorder. Bressman SB et al19 studied forty Ashkenazi patients with secondary dystonia with age of onset < 28 years and clinical features resembling primary generalized dystonia who had normal neurological examination. Only ten patients had perinatal hypoxic insult or static encephalopathy and one patient tested positive for DYT1. Maniak S et al20 analyzed one hundred thirty index patients (age 30-81 years) with focal or segmental dystonia, of which one hundred twenty had focal dystonia. Of these eighty nine (74%) had cervical dystonia, sixteen (13%) had writer’s cramp and fifteen (13%) had blepherospasm/Meige’s syndrome. Twenty (17%) had dystonic tremor. Ten cases had segmental dystonia.
Benecke R et al21 found a defect of complex-1 of mitochondrial respiratory chain proteins in primary dystonia and most of them had troticollis, however server deficiency was found in those patients who had generalized dystonia. In present study single case of dystonia due to mitochrondrial disease was suspected due to associated proximal muscle weakness, ptosis, recurrent vomiting and headache. MRI brain showed bilateral, subcortical and basal ganglia signal intensity changes on T2 weighted images. Though muscle biopsy for ragged red fibers was negative. Genetic study could not be done.
Behari M et al22 reported sporadic case of choreo-acanthocytosis in whom laryngeal dystonia and dysphagia were the dominant symptoms. In Indian literature there are not much of the reported cases of acanthocytosis. In present study one patient had evidence of neuro-acanthocytosis and presented with oromandibular dystonia.
Nemeth A et al23 reported a family in which five members over three generation had abnormal movements. Five had dystonia and three had tic disorder, thus, suggesting that these two disorders can have common genotype. In our study there was one cases of Tourette's syndrome with dystonia who presented with complex tic disorder and generalized dystonia. Family history was traced upto third generations and no other member was having tics or dystonia. Cranial imaging was normal.
Singh S et al24 reported a twenty three year old male patient who presented with two years history of torticollis with sleep benefit. Levodopa with carbidopa (100 + 25 mg) twice a day improved his torticollis completely. In present study none among seven patients with dopa responsive dystonia had cervical involvement.
Goyal V et al25 reported a thirteen year girl with ataxia telangiectasia who presented with generalized dystonia and had conjunctival telangiectasia and mental subnormality, choreoathetosis, myoclonic, jerks and bilateral cerebellar signs. Serum alfa fetoproteins, IgG and IgA level were raised. MRI showed bilateral mild cedrebellar atrophy. In present study there was one male patient of same age group with similar presentation with diagnosis of ataxia telangiectasia.
BehariM et al26 conducted a pilot case-control study for identifying possible risk factors for Meige' syndrome. They found that betalnut with tobacco chewing was a significant predictor for Meige's syndrome. The role of local irritation or the effect of some chemicals in tobacco and betal nuts need further evaluation. In present study there was a single case of Meige's syndrome in whom no cause could be found.
Das SK et al27 conducted a community based study on dystonia from India reported crude prevalence rate of primary dystonia 43.91/100,000 and age standardized rate 99.06. All cases were focal type and predominantly Writers cramp. Mean onset of dystonia was earlier in women as compared to men.
Kalita J et al28 reported clinical and MRI findings of seventeen patients with oromandibular dystonia due to Japanese encephalitis and non-specific encephalitis. Their median age was fourteen (2-53) years and nine were females. Cranial imaging was abnormal in thirteen patients and SPECT was abnormal in fifteen patients.
Tufan N et al29 reported that three novel mutations in GCH1 gene have been found and were shown to be associated with variable clinical phenotypes mostly within the spectrum of dopa responsive dystonia. Severity of clinical phenotype and the age of onset varied among family members harbouring the same mutations.
Koukouni V et al30 reported that young onset primary cervical dystonia is uncommon and in seventy six patients with young onset primary cervical dystonia mean age of onset was twenty one years. Family history of dystonia was noticed in 26.3%. Preceding injury or surgical intervention was noted in 17.1%.
Skogseid IM31 reported that the discovery of the first two gene mutations causing primary generalized dystonia [(DYT1-TORIA) and (DYT16-THAP1)] has facilitated studies on pathogenesis and pathophysiology of primary dystonia.
Brian D et al32 reported that using latest technologies in human genetics, new genetic links are being discussed and expands our knowledge of the disorders complex pathogenesis.
Fung VS et al33 reported that in sporadic isolated dystonia, the chance of finding a genetic cause in less than 1-2%, so genetic testing usually is not cost effective. The diagnostic approach in younger individuals with isolated sytonia in quite different, because there is much higher likelihood of disclosing a cause.
CONCLUSION
This study gives details of demographic and clinical features of dystonia in less than forty years age group of Indian population. This study concluded focal dystonia are more common (57.73%) than generalized dystonia. Torticollis which is considered to be most common dystonia in previous studies, was second common focal dystonia in this study. Wilson's disease was the most common cause for secondary generalized dystonia (9.09% of all dystonia).
In sixty four out of two hundred nineteen (29.22%) cases with dystonia, we were able to make an etiological diagnosis.
The limited facilities for investigations (biochemical and genetic) did not help to establish diagnosis after clinical diagnosis was made. Imaging in two cases both with brain tem AVM established the cause of dystonia. All of six patients of dopa responsive dystonia responded to small doses of levodopa (100mg/d). Hemidystonia usually occurs in the setting of a structural lesion of the contralateral basal ganglia with stoke being the commonest, followed by trauma and perinatal insults.
A systematic approach to dystonia helps to ensure that patients with this disorder receive optimum care and describe how to apply the clinical information for selection of appropriate laboratory investigations.

References:
-
Fahn S. Concept and classification of dystonia. Advances Neurol 1988; 50: 1-8.
-
Flatau S, Sterling W. Progressiver Torsconsparrybie Kundera Zeilschriftgesante fur Neurologic und Psychiatrie 1981; 7: 586-612.
-
Marsden CD. Investigation of dystonia. Advances Neurol 1988; 50:35-44.
-
Butter AG. An epidemiological survey of dystonia with the entire population of North East England over the past 9 years. Fourth International Dystonia Symposium 2002, Atlanta, Georgia, USA.
-
Fahn S, Bressman SB, Marsden CD. Classification of dystonia. Advances Neurol 1998; 78:1-10.
-
Calne DB, Lang AE. Secondary dystonia. Advances Neurol 1988; 50: 9-33.
-
Ceballos-Bauman AO, Sheean G, Passingham RE, Marsden CD, Brooks DJ. Botulinum toxin does not reverse the cortical dysfunction associated with writer’s cramp. A PET study. Brain 1997; 120: 571-582.
-
Magyar-Lehman S, Antonini A, Roelcke U, Maguire R, Missimer J, Meyer M, Leenders KL. Cerebral glucose metabolism in patients with apasmodictorticllies. Mov Disorder 1997; 12 (5): 704- 708.
-
Fahn S, Marsden CD, Calne DB. Classification and investigation of dystonia. In: Marsden CD, fahn S (eds.): Movement disorders, 2 london, Butter University pp 332-353.
-
Susan B. Bressman. Dystonia current opinion. Neurology 1998; 363-372.
-
Friedman J, Standaert DG. Dystonia and its disorders. Neurol Clin 2001;19: 681-705.
-
Fahn S. Assessment of primary dystonia. In:Munset TL, ed. Quantification of Neurologic deficit. Boston, MA: Butterworth, 1989; 241-270.
-
Nutt JF, Muenter MD, Aronson A, Kurland LT, Melton LJ III. Epidemiology of focal and generalized dystonia in Rochesters Minnesota. Mov Disorder 1988; 3; 188-194.
-
Bressman SB, de Leon D, Raymond D, et al. clinical genetic spectrum of primary dystonia. Advances Neurology 1988;78:79.
-
Ozelius LJ, Hewett JW, Page CE, et al. the early-onset torsion dystonia gene (DYT1) encodes an ATP binding protein. Nat Genet 1997; 17; 40.
-
Chuang C, Fahn S, Frucht S. The natural history and treatment of acquired hemidystonia: report of 33 cases and review of lecture. J Neurosurg psychiatry ; 72:59-67.
-
Defazio G, Berardelli A, Abbruzzese G, et al. possible risk factors for primary adult onset dystonia: a case-control investigation by the Italian movement disorders study group. J Neurosurgeon Psychiatry 1998; 64:25-32.
-
Pettigrew L, Jankovic J. Hemidystonia: report of 22 patients and a review of the literature. Journal of Neurology, Neurosurgery and Psychiatry. 1985;48: 650-657.
-
Bressman SB, Leon D,Raymud D, et al. Secondary dystonia and the DYT1 gene. Neurology 1997; 48:1571-1577.
-
Maniak S, Sieberer M, Hagenath J, et al. Focal and segmental pimary dystonia in North and Western Germany – a clinico –genetic study. Acta Neurol Scand 2003; 107: 228-232.
-
Benecke R, Strumper P, Weiss H. Electron transfer complex-1 defects in idiopathic dystonia. Ann Neurol 1992;32:686.
-
Behari M, Saha P, Prasad K, Ahuja G. Choreoacanthocytosis with marked dysphagia and laryngeal dystonia. JAPI 1991; 39(12): 967-968.
-
Nemeth A, Mills K, Elston J, et al. Do the same genes predispose to Gillis de la Touette syndrome and dystonia? Report of a new family and review of the literature. Movement Disorders 1999; 14 (5): 826- 831.
-
Sinngh S, Goyal V, Prasad K, Behari M. Cervical dystonia responsive to levodopa. Neurology India 2004; 52(2): 276-277.
-
Goyal V, Behari M. Dystonia as presenting manifestation of ataxia telangiectasia: A case report. Neurology India 2002; 50: 187-189.
-
Behari M, Sharma A, Sharma N, et al. Case-control study of Meige’s syndrome. Result of pilot study. Neuroepidemiology 2000;19:275-280.
-
Shyamal Kumar Das, Bhaskar Ghosh, Gautami Das, Arindam Biswas, Jharna Ray. Movement disorders - Indian scenario: A clinic-genetic review. Neurology India 2013; 61(5)(: 457-466).
-
Kalita J, Misra UK, Pradhan PK. Oromandibular dystonia in encephalitis. J Neurol Sci 2011 May; 304: 107-110.
-
Tufan Naiya, Amar K. Misra, Arindam Biswas, Shyamel K Das, Kunal Ray, Jharna Ray. Journal of Neural Transmission Nov. 2012; 119(11): 1343-1350.
-
Koukouni V, Markand D, Arabia G, Quinn NP, Bhatia KP. The entity of young onset primary cercical dystonia. Mov Disorder 2007 April; 22(6):843-7.
-
Skogseid IM. Dystonia – New advances in classification, genetics, pathophysiology and treatment. Acta Neurological Scandinavica April 2014; Vol. 129 (Suppl. 98): 13-19.
-
Brian D. Berman, HA Jinnah. Dystonia – Five new things. Neurology Clinical Practice June 2015; Vol. 5(3): 232-240.
-
Fung VS, Jinnah HA, Bhatia K et al. Assessment of the patient with isolated or combined dystonia: An update in dystonia syndrome. Mov Disorder 2013; 28: 889-98.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





