IJCRR - 9(23), December, 2017
Pages: 22-24
Print Article
Download XML Download PDF
A Case of Neurofibromatosis Type 1 Associated with Cervical Cord Ependymoma
Author: Ankur Mittal, Rattilal Meena, Neera Samar, Satish Kumar, Ashish Khandelwal
Category: Healthcare
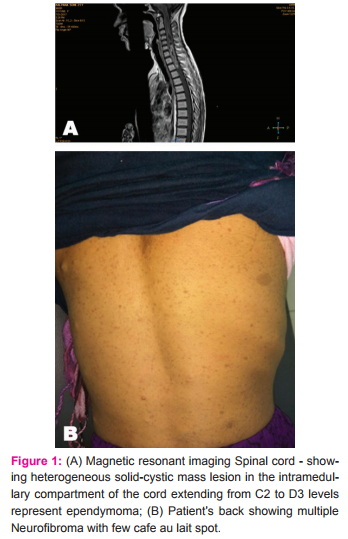
Abstract:Patients with neurofibromatosis 1 (NF1) are predisposed to develop central nervous system tumours, due to the loss of neurofibromin, an inactivator of proto-oncogene RAS. We present a case of NF1 patient with a spinal cord ependymoma. She presents with 3 months history of increasing weakness in right Upper limb and lower limb associated with numbness with involvement of left half of the body since 15 days. Magnetic resonance imaging revealed heterogeneously enhancing solid cystic mass lesion in the intramedullary compartment of the cord extending from C2-C3 to D2-D3 levels with cord edema up to medulla and D11 level craniocaudally respectively, likely to represent ependymoma.
Keywords: Autosomal dominant disorder, Ependymoma, Neurofibromatosis 1, Neurofibromin
DOI: 10.7324/IJCRR.2017.9235
Full Text:
Introduction
Neurofibromatosis 1 (NF1) is an autosomal dominant disorder caused by heterozygous mutations of the NF1 gene, which is located on chromosome 17q11.21, 2, 3. Mutations in NF1 result in loss of, an inactivator of proto-oncogene RAS, leading to increased proliferation and tumorigenesis, therefore, patients with NF1 are predisposed to develop innocent and malignant tumors4,5. In central nervous system, gliomas are the most common neoplasms in individuals with NF16, however, ependymoma with NF1 has rarely been reported. To date, only three cases have been reported in English literature7,8. Moreover, cervical spinal cord ependymoma, to the best of our knowledge, has never been reported occurring in NF1 patients previously. Recently, we experienced a case of cervical spinal cord ependymoma in a patient with NF1. In this report, we discuss the diagnosis, the clinical management, mechanisms of such a rare case with review of the literature.
Case report
A 21 Year old female patient was admitted to our department for the complaint of weakness of right half of the body since 3 months which was associated with numbness in right half of the body later on involving the left half of the body since 15 days. In her past medical history there were expeditiously increscent cutaneous neurofibromas respectively on her back, lower limb and upper limb which were present since childhood. In her family history, her mother and her maternal grandmother has the similar cutaneous neurofibromas for which they never consulted a doctor and also doesn’t have any neurological complaint in their lifetime. On physical examination, widespread café-au-lait spots, axillary and groin frecklings, cutaneous neurofibromas. No iris hamartomas had been found and mammary gland was normal. In neurological examination, her mental functions were normal, speech normal, all cranial nerve were normal, motor system examination reveals increased tone in both lower limbs, power was 4/5 in all four limbs, Deep tendon reflexes were absent in both upper limb and exaggerated in both lower limb. Planters were bilaterally extensor. Sensory system reveals diminished sensation for all the primary modalities of sensation. No neck rigidity. No cerebellar signs.
In RNT medical college all the investigations were performed. In which Haemoglobin- 13.5gm%; White blood count -8510/mcl; Platelets- 3.83 lacs/mcl; PBF-Normocytic normochromic; ESR- 29; Blood glucose- 103mg/dl; Urea-25mg/dl; creatinine-0.55mg/dl; liver function tests were normal; Lipid profile was normal; Serum electrolyte(Sodium=137; Potassium=4.6; Chloride=108; Calcium=8.7); Chest X-Ray Posterioanterior view and Electrocardiogram were normal; Ultrasonography abdomen does not show any significant abnormalities; Fundus examination was normal.
Magnetic resonant imaging study reveals heterogeneous solid-cystic mass lesion in the intramedullary compartment of the cord extending from C2-C3 to D2-D3 levels appears iso-intense on T1 weighted & heterogeneously hyper intense on T2 weighted sequence. There is heterogeneous enhancement on the post contrast study with non enhancing cystic component. There is cord edema craniocaudally up to medulla superiorly and D11 inferiorly appears isointense on T1 weighted and hyper intense on T2 weighted sequence represent ependymoma.
Magnetic resonant imaging brain doesn’t show any significant abnormality.
Discussion
Neurofibromatosis-1(NF1) occurs with approximately 1: 2000 to 1: 5000 in individuals3. The diagnosis of most NF1 patients is based on clinical manifestations. Diagnosis requires at least two major criteria - 2 or more neurofibromas or 1 plexiform neurofibroma, 6 or more café-au-lait patches, axillary or groin freckling, optic pathway glioma, lisch nodules in the Iris, a distinctive osseous lesion, a first degree relative with NF1. The clinical manifestations of our patient revealed a typical NF1.
Spinal cord ependymomas are the most common intramedullary tumours in adults which account for 60% in all spinal cord tumours, and cervical region are the most common localization they occur9,10,11. The clinical course of our patient is consistent with spinal cord ependymoma.
Gliomas are often associated with NF1, most with a low grade, mainly locate in the optic nerve12,13, and only 1% in the spinal cord12. The incidence of intramedullary gliomas in NF1 patients may be far more than their sporadic counterparts according to similar works14. Meanwhile, compared with NF215, 16,17), ependymomas were reported rarely to occur in NF1 patient so our patient is a rare case presented with manifestations mentioned above
The clinical courses of our patient and others revealed that there were no abnormality between ependymomas in NF1 patients and their sporadic counterparts. Concurrent NF1 glioma mechanism and NF1 genes may closely relate. This gene locates on the long arm of chromosome 17 in the area of 17q11.2, can encode and achieve the synthesis of. This protein as proto-oncogene RAS inhibitors, when NF1 gene function deficiency can result in tumour formation. In addition, NF1 germline mutation, can also lead to tumour form5,11, 18). However, specific mechanisms for NF1 complicated with glioma are still unclear, molecular mechanism of tumour gene and potential therapeutic targets for tumour may be becoming the trend and direction of research 4,18,19.
Conclusion
Ependymoma with NF1 is a rare situation, we first report a spinal cord ependymoma which occurred in a NF1 patient. Referencing other cases, we find that the clinical course of ependymomas in NF1 patients have no abnormality compared with their sporadic counterparts. We emphasize that a detailed history talking and physical examination to a NF1 patient are needed and a multidisciplinary cooperation is essential. Further elucidation on the molecular changes of NF1 that drive tumorigeness remains needed aiming to explore a potential therapeutic protocol.
References:
1. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12:1–11.
2. Lakkis MM, Tennekoon GI. Neurofibromatosis type 1. I. General overview. J Neurosci Res.2000;62:755–763.
3. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33–40.
4. Brems H, Beert E, de Ravel T, Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10:508–515.
5. Cnossen MH, van der Est MN, Breuning MH, van Asperen CJ, Breslau-Siderius EJ, van der Ploeg AT, et al. Deletions spanning the neurofibromatosis type 1 gene : implications for genotype-phenotype correlations in neurofibromatosis type 1? Hum Mutat. 1997;9:458–464.
6. Rodriguez FJ, Perry A, Gutmann DH, O'Neill BP, Leonard J, Bryant S, et al. Gliomas in neurofibromatosis type 1 : a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol.2008;67:240–249.
7. Riffaud L, Vinchon M, Ragragui O, Delestret I, Ruchoux MM, Dhellemmes P. Hemispheric cerebral gliomas in children with NF1 : arguments for a long-term follow-up. Childs Nerv Syst. 2002;18:43–47.
8. Sharma AS, Emery ME, Metcalfe KM, Sabin HIS, Drake WMD. A case of thoracic cord ependymoma in neurofibromatosis type 1. Endocr Abstr. 2006;12:17.
9. Alkhani A, Blooshi M, Hassounah M. Outcome of surgery for intramedullary spinal ependymoma. Ann Saudi Med. 2008;28:109–113.
10. Eroes CA, Zausinger S, Kreth FW, Goldbrunner R, Tonn JC. Intramedullary low grade astrocytoma and ependymoma. Surgical results and predicting factors for clinical outcome. Acta Neurochir (Wien)2010;152:611–618.
11. Son DW, Song GS, Han IH, Choi BK. Primary extramedullary ependymoma of the cervical spine : case report and review of the literature. J Korean Neurosurg Soc. 2011;50:57–59.
12. Guillamo JS, Créange A, Kalifa C, Grill J, Rodriguez D, Doz F, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1) : a retrospective study of 104 patients. Brain. 2003;126(Pt 1):152–160
13. Rosenfeld A, Listernick R, Charrow J, Goldman S. Neurofibromatosis type 1 and high-grade tumours of the central nervous system. Childs Nerv Syst. 2010;26:663–667.
14. Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1 : an analysis using U.S. death certificates. Am J Hum Genet. 2001;68:1110–1118.
15. Aguilera DG, Mazewski C, Schniederjan MJ, Leong T, Boydston W, Macdonald TJ. Neurofibromatosis-2 and spinal cord ependymomas : report of two cases and review of the literature. Childs Nerv Syst.2011;27:757–764.
16. Lim BS, Park SQ, Chang UK, Kim MS. Spinal cord tanycytic ependymoma associated with neurofibromatosis type 2. J Clin Neurosci. 2010;17:922–924.
17. Ueki K, Sasaki T, Ishida T, Kirino T. Spinal tanycytic ependymoma associated with neurofibromatosis type 2--case report. Neurol Med Chir (Tokyo) 2001;41:513–516.
18. Graf N. Glioblastoma in children with NF1 : the need for basic research. Pediatr Blood Cancer.2010;54:870–871.
19. Dilworth JT, Kraniak JM, Wojtkowiak JW, Gibbs RA, Borch RF, Tainsky MA, et al. Molecular targets for emerging anti-tumour therapies for neurofibromatosis type 1. Biochem Pharmacol. 2006;72:1485–1492.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





