IJCRR - 9(19), October, 2017
Pages: 26-31
Print Article
Download XML Download PDF
Prediction of Potential Lead Molecules through Systematic Integration of Multi-omics Datasets - A Mini-Review
Author: Ashok Kumar T., Rajagopal B.
Category: General Sciences
Abstract:Prediction of a novel or potential lead molecules for a therapeutic drug target without adverse effects is a challenging task in the drug designing, discovery, and development process. The systematic integration of multi-omics data from various data/knowledge bases through computational techniques enables to identify potential lead molecules and study the therapeutic properties. Over the last decades, several drug discoveries using multi-omics and huge dataset integration methods proven with successive results. In this paper, we present different types of computational approaches for prediction of potential lead molecules through the systems-level integration of multi-omics datasets.
Keywords: Systematic Integration, Multi-omics Datasets, Drug Discovery, Lead Identification, Big Data Analysis
Full Text:
INTRODUCTION
In drug discovery, lead is a chemical compound that binds to active site regions of the biological target molecule and hence minimizes the binding free energy. Leads may be a natural product, synthetic, or semi-synthetic compound which has therapeutic effects[1].Natural product (or natural drug) consists of bioactive compounds which were produced by the living organisms that are present in nature. Plants, minerals, and animals (including microorganisms) are the common sources of natural products[2,3,69,74,75]. Natural products can also be developed by chemical synthesis (both semi-synthesis and total synthesis) and have been placed a major role in the development of potential synthetic targets. But synthetic and semi-synthetic compounds are chemically synthesized by the humans in the laboratory using in silico and/or experimental approaches[4,5].
Developing a potential lead molecule by using the experimental method is tedious, complicated, expensive, time-consuming, and trial-and-error process[6]. Recently, many advanced computational techniques analogous to wet-lab techniques were introduced to reduce the problem. Modern computer-aided drug design and discovery (CADDD) involve virtual screening, testing, and validation of lead molecules in a short time span using large datasets and software [73].The resulting lead molecule further undergoes a series of preclinical and clinical studies to test the toxicity and adverse effects. The successful drug candidate is released in different dosage forms in the market after passing the food and drug administration (FDA) verification process[7,8].
MULTI-OMICS AND BIG DATA INTEGRATION
Multi-omics is a new approach for analyzing biological problems in various aspects through combining multipleomics datasets[3,9]. The common types of omics include genomics, proteomics, metabolomics, epigenomics, phytochemomics, interactomics, and microbiomics[10-12]. Integration of multiple omics data in a systematic way enables to study the functional relationship or identify the key problem in an efficient manner. An association of large datasets or complex datasets of multi-omics data is a difficult task and must have sound knowledge in all areas of omics.The pattern matching (or regular expression) is a general and most popular technique for extraction of knowledge from the datasets. Analyzing the large multi-omics datasets involves big data handling.
Due to rapid growth in data size, diversity, and complexity of datasets in the biological databases, big data were introduced to analyze, manage, and derive knowledge from the datasets. Big data (aka huge data or massive data) refer to a very large volume of data or data storage, which cannot be processed using traditional computing devices and applications. Size of big data ranges from petabytes(1 PB = 1015 bytes) to exabytes (1 EB = 1018 bytes), or even more[13-15].Even though the big data analysis is a hot topic today, the concept has evolved over many years ago in IT and R and D sector. Next-generation sequencing (NGS) and drug discovery are the two most popular areas of biological sciences which currently implement big data analysis in knowledge discovery[16-18].
Comprehensive Data Integration Methods
Integrating comprehensive and related datasets from various biological databases or other external sources increases the accuracy in lead prediction, and also reveals hidden functions and interrelationship within the molecules[19].There are three types of approaches adopted to combine comprehensive data and reduce data size (Table 1): (i) semantic web approach - searching, retrieving, or annotating data from other external data sources through metadata or a RESTful APIweb services [20,21]; (ii) data warehousing approach - extracting data from other external sources and combining into a global dataset[19,22]; and (iii) data mining approach - extracting data or knowledge from different types of large datasets through suitable patterns[23,24].
Most of the popular three-dimensional (3D) molecular structure databases such as RCSB Protein Data Bank[25], NCBI PubChem[26], EMBL-EBI ChEBI [27], Drug Bank[28], etc. have implemented REST ful API web services or SOAP to share or integrate data in the form of FTP, HTML, XML, JSON, plain text, or AWK commands[29].Moreover, cloud computing services were offered to handle, analyze, or interpret big datasets through various remote applications/servers. There are many cloud servers such as Cloud BLAST[30], Myrna[31], Cloud Burst[32], Hadoop-BAM[33], GPU-BLAST[34], Hydra[35], Peak Ranger[36],Crossbow[37], etc. were available over cloud for analyzing different types of big datasets [38-41].
Unsupervised Data Analysis and Analytics
Handling big dataset or multi-omics data is a difficult task, because it is often very comprehensive and available in real time. In Bioinformatics, sequence (alphabets) and structure (XYZ coordinates) are the major data used for big data analysis and analytics. An effective lead identification and functional interrelationship prediction require integration of very large datasets of3D chemical libraries and disease-target-ligand interaction network. Usually unsupervised multi-omics/big datasets are integrated using clustering and grouping technique. The different types of dataset integrations are target-ligand interactions, intermolecular interactions, disease-target interactions, disease-disease relationships, protein-protein interactions, target-disease-metabolic pathways, drug-side effect relationships, gene interactions, structure-function relationships, etc. [42-44].
The network model graphical representation of biological data interrelations and various types of unsupervised dataset integration methods are [44,56]:(i) network-based methods - graphical representation of interrelations using the network (distance) datasets [45,46],(ii) Bayesian methods - probabilistic graphical representation of interrelations using the probability distribution datasets [47-51], (iii) correlation-based methods - multivariate graphical representation of interrelations using the partial least squares datasets [52,53], (iv) matrix factorization methods - graphical representation of interrelations using the product and rank of the two matrix datasets [54], and (v) kernel-based methods - graphical representation of interrelations using the pattern datasets predicted from kernel matrix [55].
Big Data Accessing Methods
Accessing large datasets requires high-performance computing (HPC) infrastructure and a suitable big data framework [14,15]. The common methods for big data handling are cloud computing, graphics processing unit (GPU) computing, Xeon Phi computing, grid computing, and cluster computing [57,58]. Large datasets can be accessed from various data sources using big data framework, which is based on client-server technology [59]. There are many types of big data processing frameworks used for accessing datasets through a pipeline, among which popularly used frameworks and programs are: Apache Hadoop [76], Apache Spark [77], Apache Flink [78], Apache Storm [79], Apache Samza [80], Apache Cassandra [81], NoSQL [82], R [83], and Python[84].
SYSTEMATIC MULTI-OMICS DATA INTEGRATIONA successful drug discovery requires exact compound or most suitable compound which can fit all pocketsin the active site of the target molecule and brought to a stable state [7,8]. The systematic integration of theoretical and experimental datasets of multi-omics, target-ligand interaction network, physicochemical properties, and functional properties leads to design a safe and efficient therapeutics [60].
Integrative Systems Biology Approach
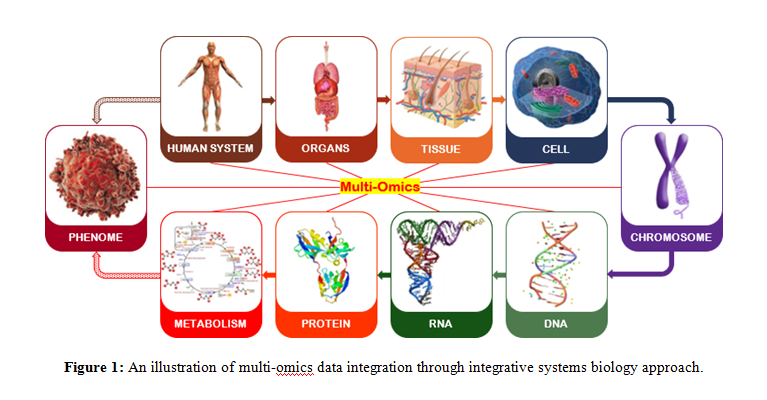
To design an effective drug molecule, it is most essential to understand the nature and causes of the disease [61].Integrative systems biology advances thorough study of biological phenomenon of a system (organism, e.g. human) in a systematic way (Figure 1).The complex interaction networks in a system can be combined through either top-down or bottom-up approaches using multi-omics datasets [62,63].Currently there are many bioinformatics databases and tools were available for collection of various omics data and hence can design a new virtual system.
Computational Methods for Lead Identification
A lead molecule can be identified by integrating or comparing target data with large datasets using computational and statistical approaches. The common computational lead identification techniques using large datasets include:
- Multiple sequence alignment -It is a popular method to find local similarity, homology, and phylogenetic relationship between different genes or protein sequences [41]. The sequence similarity through structure-based sequence alignment enables to find the similar target-ligand interacting molecules. Structural superposition is another alternative approach to compare similar protein structures based on the root mean square deviation (RMSD) calculation [64]. Moreover, systematic integration of large datasets of target-ligand molecular interaction network data with multi-omics data enables to predict or design a potential lead molecule [60].
- Maximum common substructure - It is a widely used method in CADD for finding similar 3D structures through structured-based or ligand-based virtual screening [60].Maximum common substructure search using SMILES (Simplified Molecular Input Line Entry System)pattern is commonly used to find structural similarity between large chemical datasets [65].The substructure search with compounds in the phenotype linked target-ligand interacting network datasets integrated with multi-omics data enables to predict or design a novel and potential lead molecule [66-68].
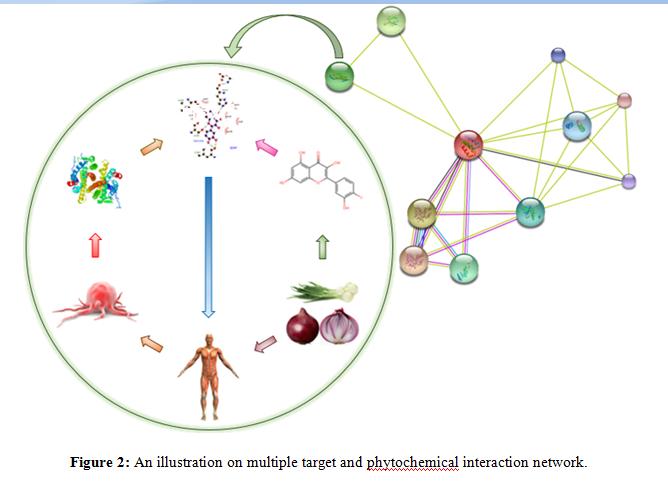
- Molecular interaction network - It is the modern and most successive approach to find a novel drug by systematic integration of large datasets of multi-omics data [60].Data scientists integrates big data into complex network in the order of phenotype ? target ?target-ligand?ligand?chemical library to predict or design a novel and potential lead molecule (Figure 2). Recently, many big pharmaceutical companies and R and D organizations have renewed their interest in discovering potential lead compounds from the natural products due to the structural diversity and medicinal properties [3,69,70].
CONCLUSION
Biological systems are analogous to the computer system in disease/target identification and drug design. To troubleshoot hardware issues in the computer, we must have the complete circuit diagram and the component to fix the problem [71]. In contrast, through increasing the volume of multi-omics datasets and systematic integration of large datasets, it is possible to design an effective drug molecule [72]. Recent research advances in cloud computing, big data analysis, multi-omics data integration, and virtual screening and testing technology have reduced the cost and time in predicting potential lead molecules.
ACKNOWLEDGEMENT
Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to authors / editors / publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed.
Conflict of Interest
The authors declare that there is no conflict of interest regarding publication of the paper.
Table 1: A comparison between various comprehensive data integration approaches.
|
Approaches
|
Advantages
|
Disadvantages
|
|
Semantic web
|
- Occupies less storage space
- Provides more information
- Provides updated information
- High quality of data
- Multiple access options
|
- Non-uniform data from external sources
- Sometimes links may be broken
- The data access format may be changed
- Sometimes data may be ambiguous
- Interlinking is not possible
- Sometimes data process may timeout
- Interrelationship study is not possible
|
|
Data warehousing
|
- Provides more information
- High quality of data
- Uniform access options
- Interlinked to the target source
- Can predict interrelationships
- Can add more features
|
- Occupies more storage space
- Provides outdated information
- Manual data synchronization
|
|
Data mining
|
- Provides updated information
- Uniform access options
- Interlinked to the target source
- Can predict interrelationships
- Can add more features
|
- Occupies more storage space
- Provides less information
- Less quality of data
|
References:
1. S.Z. Tasker, P.J. Hergenrother, Natural products: Taming reactive benzynes, Nat. Chem. 9 (2017) 504–506.
2. M. Lahlou, Screening of natural products for drug discovery, Expert Opin. Drug Discov. 2 (2007) 697–705.
3. T. Ashok Kumar, B. Rajagopal, PDTDB – An Integrative Structural Database and Prediction Server for Plant Metabolites and Therapeutic Drug Targets, Int. J. Curr. Res. 9(2017) 46537– 46541.
4. All natural, Nat. Chem. Biol. 3 (2007) 351–351. 5. A.M. Lourenço, L.M. Ferreira, P.S. Branco, Molecules of natural origin, semi-synthesis and synthesis with anti-inflammatory and anticancer utilities, Curr. Pharm. Des. 18 (2012) 3979–4046. 6. F. Ooms, Molecular modeling and computer aided drug design. Examples of their applications in medicinal chemistry, Curr. Med. Chem. 7 (2000) 141–158. 7. I.M. Kapetanovic, Computer-aided Drug Discovery and Development (CADDD): in silico-chemico-biological approach, Chem. Biol. Interact. 171 (2008) 165–176. 8. G. Sliwoski, S. Kothiwale, J. Meiler, E.W. Lowe, Computational Methods in Drug Discovery, Pharmacol. Rev. 66 (2013) 334–395. 9. A. Ebrahim, E. Brunk, J. Tan, E.J. O’Brien, D. Kim, R. Szubin, J.A. Lerman, A. Lechner, A. Sastry, A. Bordbar, A.M. Feist, B.O. Palsson, Multi-omic data integration enables discovery of hidden biological regularities, Nat. Commun. 7 (2016) 13091. 10. M. Bersanelli, E. Mosca, D. Remondini, E. Giampieri, C. Sala, G. Castellani, L. Milanesi, Methods for the integration of multi-omics data: mathematical aspects, BMC Bioinformatics. 17 (2016) 167–202. 11. C. Bock, M. Farlik, N.C. Sheffield, Multi-Omics of Single Cells: Strategies and Applications, Trends in Biotechnol. 34 (2016) 605–608. 12. C. Vilanova, M. Porcar, Are multi-omics enough?, Nat. Microbiol. 1 (2016) 16101. 13. M. Swan, The Quantified Self: Fundamental Disruption in Big Data Science and Biological Discovery, Big Data. 1 (2013)
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





