IJCRR - 1(2), November, 2009
Pages: 38-43
Print Article
Download XML Download PDF
SYNTHESIS AND ANTITUBERCULAR ACTIVITY OF SOME NEW BENZOPYRONE DERIVATIVES
Author: Shashikant R.Pattan, Nachiket s Dighe, Jayshri S Pattan, Santosh R Butle, Santosh G Jadhav, Deepak S Musmade Suwarna H Kale
Category: Healthcare
Abstract:A series of 5-Bromo- 2-formyl phenoxyacetyl amino acids and peptides have been synthesized by coupling of the 5-Bromo- 2-formyl phenoxyacetic acid with amino acid/methyl esters/dipeptides/ tripeptides using DCC as coupling agent and NMM as base. The structures were elucidated FTIR and 1HNMR .The newly synthesized compounds were evaluated for their antibacterial, antifungal and anthelminitic activities. The compounds (2, 6, 11 and 13) were found to exihibit potent antibacterial activity against Bacillus subtilis, Staphylococcus aureus (gram positive bacteria) and Escherichia coli (gram negative) bacterias.The compounds (5, 6 and 13) were found to exihibit potent antifungal activity against Candida albicans and Aspergillus niger. The moderate to good anthelminitic activity was shown by the synthesized compounds (7 and 13) against Eudrilus spieces.
Keywords: Phenoxyacetic acid, amino acids, antibacterial, antifungal and anthelminitic.
Full Text:
Phenoxyacetic acid is among the most vital moieties which are associate with potent antidiabetic (Rival et al 2004), antimycobacterial (Yar et al 2007),diuretic (Lebedev et al 1985, Woltersdorf et al 1976, Bicking et al 1976), anti-inflammatory (Kunsch et al 2005, Shokol et al 2005), antibiotic (Grardin et al 1995), anti-obesity (Kiso et al 1999), diagnostic (Ohmomo et al 1989), inhibition of platelet aggregation (Meanwell et al 1993, Seiler et al 1994) activities. The review of literature has suggested that incorporation of amino acids and peptides into aromatic and heterocyclic congeners have resulted in compounds with potent bioactivities.
Introducing an amino acid or peptide into aromatic compounds can increase the potency, decrease the toxicity and prolong its action. Among aromatics, phenolic compounds have wide range of activities. Further phenoxylation the resulting compound phenoxyacetic acid is obtained, which is well known for their biological potential. Thus keeping in view the biological potency of phenoxyacetic acids as well as taking advantage of biodegradability and biocompatibility of a novel series of substituted phenoxyacetic acid derivatives of amino acids and peptides have been synthesized with an anticipation to get potent agents with good therapeutic efficacy with negligible side effects. The study of IR and 1HNMR spectrum gives us most of the required information of certain vibrational bands or characteristic groups present in the molecule. The IR spectrum of newly synthesized compounds showed characteristic bands in the region 3329- 3318, 1654-1642, 1537-1532 cm-1 which can be assigned as N-H stretching, C=O stretching and N-H bend respectively. In their 1HNMR spectra, a singlet appeared in the range 8.33-8.36 ppm corresponding to the CO-NH proton. The compounds were found to exhibit potent antimicrobial and moderate anthelmintic activity in comparison to standard drugs against the same concentration.
Results and discussion
Antibacterial Activity:
The synthesized peptide derivatives were screened for antibacterial activity against Escherichia coli, Staphylococcus aureus and Bacillus subtilis using modified Kirby-Bauer disc diffusion method (DMF as a solvent).The test samples were tested at the concentrations 25, 50, 100 pg/ml. The petri plates inoculated with bacterial cultures were incubated at 37C for 18 hrs. The diameters obtained for the test sample were compared with that produced by the standard drug ciprofloxacin. The results are shown in Table 2.
Antifungal Activity:
The synthesized peptide derivatives were screened for antifungal activity against Candida albicans and Asperigillus niger. DMSO is used as negative control. The test samples were tested at the concentrations 25, 50, 100 pg/ml. The petri plates inoculated with fungal cultures were incubated at 25 C for 48 hrs. Diameters of the zone of inhibition were calculated in triplicate sets. The diameters obtained for the test sample were compared with that produced by the standard drug griseofulvin. The results are shown in Table 2.
Anthelmintic Activity:
The anthelminitic activity was carried out against earthworms Eudrilus species by Garg and Atal method at 2 mg/ ml concentration. Suspension of samples was prepared by triturating synthesized cyclic peptide (200 mg) with Tween 80 (0.5 %) and distilled water. Suspension of the standard drug albendazole was prepared with the same concentration in a similar way. The paralyzing and death times were noted and their mean was calculated for triplicate sets. The death time was ascertained by placing the earthworms in warm water (50C) which stimulated the movement. The results were shown in Table 3. The results of biological activities revealed that newly synthesized peptide derivative 73 at 50pg/ml concentration exhibited highest zone of inhibition against Staphylococcus aureus and 75 at 50pg/ml concentration exhibited highest zone of inhibition against Candida albicans. Morover, other compounds showed moderate antimicrobial activities against tested organisms. Comparison of anthelmintic activity data revealed that peptide derivative 75 was found to exhibit potent anthelmintic activity and other peptide derivatives showed good to moderate activity.
Experimental
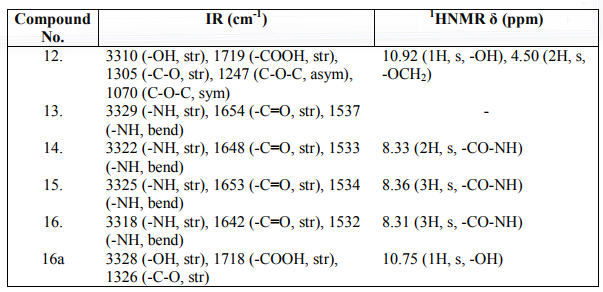
Melting points were determined and uncorrected. The amino acids, di-tertbutyl pyrocarbonate (Boc2O), 5- Bromosalicyaldehyde, DCC and NMM were obtained from Spectrochem Limited, Himedia laboratories Limited mumbai and Sd-fine-chem Limited, Mumbai, India. The IR spectra were recorded on a Perkin Elmer Fourier transform infrared spectrophotometer using KBr pellets. The 1HNMR spectra were recorded on the Bruker Avance II- 400 NMR spectrometer using CDCl3 as the solvent. The purity of all the compounds was controlled by TLC on silica gel G plates. Chloroform:Methanol (9:1 v/v) was used as developing solvent system and dark brown spots were detected on exposure to iodine vapours in a tightly closed chamber. The physical data of synthesized compounds is listed in Table 1. The scheme of synthesis is given in Scheme 1.
Synthesis of Boc amino acids (1-3):
L-Leucine (1.31gm, 10mmol) was dissolved in 10 ml of sodium hydroxide (1 mol L-1 ) and 10 ml of i-propanol. ditert.butylpyrocarbonate (3 ml, 13 mmol) in 5 ml of i-propanol was added followed by 10 ml of sodium hydroxide (1 mol L-1 ) to the resulting solution. The solution was stirred at room temperature for 2 hr, washed with 10 ml of light petroleum ether (b.p. 40-60 C), acidified to p H 3.0 with 1 mol L-1 sulphuric acid and finally extracted with chloroform (3 x 20 ml). The organic layer was dried over anhydrous sodium sulphate and evaporated under reduced pressure to give crude product. The crude product was purified by recrystallization from methanol and ether at 0 C to get pure Boc-Leucine (1). Similarly, Boc-Serine (2) and Boc-Alanine (3) were prepared by stirring di-tert.butylpyrocarbonate (3 ml, 13 mmol) with Boc-Serine (1.05gm, 10 mmol) and Boc-Alanine (0.89gm, 10 mmol) respectively.
Synthesis of L-amino acid methyl ester hydrochlorides (4-6) :
Thionyl chloride (0.73mL, 10 mmol) was slowly added to methanol (50 mL) at 0 C and 1.15 gm of L- Proline (10 mmol) was added to the above solution. The resulting mixture was refluxed for 9 hrs at 110 C. Methanol was evaporated and the residue was triturated with ether at 0 C until excess dimethyl sulphite was removed. The crude product was purified by recrystallization from methanol and ether at 0 C to get Lproline methyl ester hydrochloride (4). Similarly, L-leucine methyl ester hydrochloride (5) and L-tryptophan methyl ester hydrochloride (6) was prepared by refluxing 1.31 gm of Lleucine (10 mmol) and 2.04 gm of Ltryptophan with 50 ml methanol in the presence of 0.73 ml of thionyl chloride (10 mmol).
Synthesis of Boc-dipeptide methyl esters (7-9) :
To a mixture of 1.65 gm compound 4 (10 mmol) in 20 ml of chloroform, 2.3 ml of N- methylmorpholine (21mmol) was added at 0 C. The reaction mixture was stirred for 15 min. 2.31gm compound 1 (10mmol) in 20 ml chloroform and 2.1gm of DCC (10mmol) were added under stirring to the above mixture. After 36 hrs, the reaction mixture was filtered and the residue was washed with 30 ml of chloroform and added to the filterate. The filterate was washed with 5% sodium hydrogen carbonate and saturated sodium chloride solution (25 ml each). The organic layer was dried over anhydrous sodium sulphate, filtered and evaporated in vacuum. The crude product was recrystallized from mixture of chloroform and petroleum ether (b.p. 40-60 C) followed by cooling at 0 C to get Boc-Leu-Pro-OMe (7). Similarly Boc-Ser-Leu-OMe (8) and Boc-Ala-ProOMe (9) were prepared by stirring compounds 2 and 3 with amino acid methyl ester hydrochlorides 5 and 4, respectively in the presence of DCC and NMM.
Deprotection of dipeptides at carboxyl end (8a, 9a) :
To a solution of 3.32 gm of compound 8 (10 mmol) in 36 ml of THF/H2O (1:1), 0.36 gm lithium hydroxide (15mmol) was added at 0 C. The mixture was stirred at room temperature for 1 hr, and acidified to 1 pH 3.5 with 0.5 mol LH2SO4. The aqueous layer was extracted with diethyl ether (3 x 25 ml). Combined organic extracts were dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was recrystallized from methanol and ether to get Boc-SerLeu-OH (8a). Similarly compound 9 was hydrolyzed under alkaline conditions to obtain Boc-Ala-Pro-OH (9a).
Deprotection of dipeptide at amino end (7a):
Compound 7 (3.42 gm, 10mmol) was dissolved in 15 ml of chloroform and treated with 2.28 gm of trifluoroacetic acid (20 mmol). The resulting solution was stirred at room temperature for 1 hr and washed with 25 ml of saturated sodium hydrogen carbonate solution. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by recrystallization from mixture of chloroform and light petroleum ether (b.p. 40-60 C) to get pure Leu-Pro-OMe (7a).
Synthesis of Boc-tripeptide methyl esters (10, 11) :
To synthesize Boc-Ser-Leu-Trp-OMe (10), 3.18 gm of dipeptide unit 8a (10 mmol) was coupled with 2.54 gm of amino acid methyl ester hydrochloride 6 (10 mmol) in the presence of DCC and NMM following the same procedure as adopted for the synthesis of Bocdipeptide methyl esters 7-9. Similarly Boc-Ala-Pro-Pro-OMe (11) was prepared by coupling 2.86 gm of deprotected dipeptide unit 9a and 1.65 gm of amino acid methyl ester hydrochloride 4 using DCC as the coupling agent and NMM as the base.
Deprotection of tripeptides at amino end (10a, 11a):
Compound 10 (5.18 gm, 10mmol) was dissolved in 15 ml of chloroform and treated with 2.28 gm of trifluoroacetic acid (20 mmol). The resulting solution was stirred at room temperature for 1 hr and washed with 25 ml of saturated sodium hydrogen carbonate solution. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by recrystallization from mixture of chloroform and light petroleum ether (b.p. 40-60 C) to get pure Ser-Leu-TrpOMe (10a). Similarly Ala-Pro-Pro-OMe (11a) was prepared by stirring compound 11 with 2.28 gm of trifluoroacetic acid (20 mmol).
Synthesis of free acid
Sodium hydroxide (0.89gm, 22.4mmol) in 25 ml water was slowly added with stirring to 2.01gm of 5-bromo-2- hydroxyaldehyde (10mmol) and 0.94gm of chloroacetic acid (10mmol). The mixture was heated on heating mantle to remove all the liquid and the residue was treated with 30 ml water. The mixture was cooled and filtered and clear solution was acidified with dilute hydrochloric acid. The aqueous layer was extracted with diethyl ether (2 x 25 ml). Combined organic extracts were dried over anhydrous sodium sulphate.The crude product was recrystallized from ethanol and purified by recrystallization from ethanol-water (1:1) to get 5-Bromo-2-formylphenoxyacetic acid (12).
Synthesis of 5-Bromo-2-formylphenoxyacetyl amino acid and peptide methyl esters
L-Proline methyl ester hydrochloride (1.65 gm, 10mmol) was dissolved in Tetrahydrofuran (75 mL). To this, 2.3 ml of N-methylmorpholine (21mmol) was added at 0 C and the reaction mixture was stirred for 15 min. 2.01gm of compound 12 (10mmol) in tetrahydrofuran and 2.1gm dicyclohexylcarbodiimide (10mmol) were added under stirring to the above mixture. After 36 hrs, the reaction mixture was filtered and the residue was washed with 30 ml of tetrahydrofuran and added to the filterate. The filterate was washed with 5% NaHCO3 and saturated NaCl solution (25 ml each). The organic layer was dried over anhydrous sodium sulphate, filtered and evaporated in vacuum. The crude product was recrystallized from mixture of chloroform and n-hexane followed by cooling at 0 C to get 5-Bromo-2-formylphenoxyacetyl-proline methyl ester (13). Similarly, 5-Bromo-2-formylphenoxyacetyl-leucyl-proline methyl ester (14), 5-Bromo-2-formylphenoxyacetyl-seryl-leucyl-tryptophan methyl ester (15) and 5-Bromo-2- formyl-phenoxyacetyl-alanyl-prolylproline methyl ester (16) were prepared by stirring compound 7a, 10a and 11a with compound 12 respectively in the presence of DCC and NMM.
Deprotection of 5-Bromo-2-formylphenoxyacetyl-alanyl-prolyl-proline methyl ester at carboxyl end (16a) :
To a solution of 4.39 gm of compound 16 (10 mmol) in 36 ml of THF/H2O (1:1), 0.36 gm lithium hydroxide (15mmol) was added at 0 C. The mixture was stirred at room temperature for 1 hr, and acidified to 1 pH 3.5 with 0.5 mol L-H2SO4. The aqueous layer was extracted with diethyl ether (3 x 25 ml). Combined organic extracts were dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was recrystallized from methanol and ether to get 5- Bromo-2-formyl-phenoxyacetyl-alanylprolyl-proline (16a).
ACKNOWLEDGEMENTS
The authors are thankful to the Head, Department of Chemistry for providing research facilities, SAIF Department, Punjab University, Chandigarh (India), for providing spectral details in time.
Table 1 : Physical data of the synthesized compounds
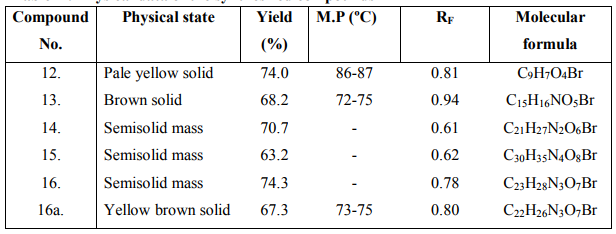
Table 2: Antimicrobial activity data of synthesized compounds
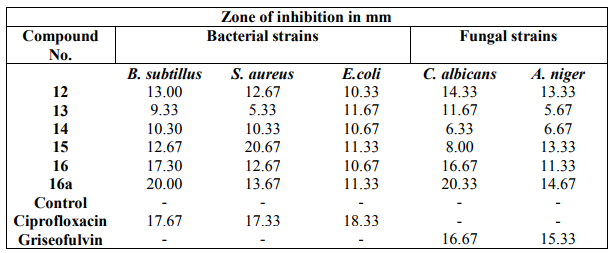
Table 3: Anthelmintic activity data of synthesized compounds
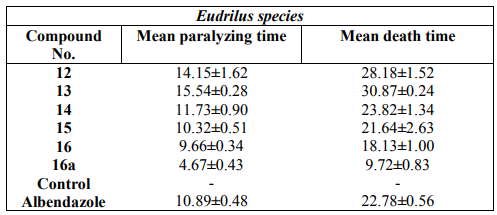
Table 4: Spectral data of synthesized compounds
References:
1. Y. Rival., A. Stennvin., L. Puech, A.Rouquete, C.Cathala, F. Lestienna,. and D.Junquero, J. Pharmacol. Exp. Therap., 2004, 311,
2, 467-475. 2. M.S Yar., A.A. Siddiqui and M.A. Ali, Bioorg. Med. Chem., 2007, 14, 4571-4574.
3. A.A. Lebedev, V.A. Smirnoc, M. Bazhmina and Karpacheva T.A., Pharm. Chem. J., 1985, 19, 3, 171- 173.
4. C. Kunsch, J. Luchoomul, G.L. Dodd, K.S. Karu, J.D. Piper and C.L. Sundell, J. Pharmcol. Exp. Therap., 2005 313, 2, 492-501.
5. P. Gerardin, M. Ahrach, R. Schneider, F. Houillon, B. Loubinoux, M. Sprenge and J.L. Colin, Bioorg. Med. Chem. Lett., 1995, 5, 14, 1467-1470.
6. T. Kiso, T. Kakita, T. Shoqaki and Y. Ohtsubo, Bio. Pharm. Bull., 1999, 22, 10, 1073-1078.
7. Y. Ohmomo, S. Okuyama, Y. Magata, Y. Ueno, C. Tanaka and A. Yokoyama, Chem. Pharm. Bull., 1989, 37, 9, 2276-2281.
8. N.A Meanwell, M.J. Rosenfeld, J.J. Kim Wright, C.L. Brassard, J.O. Buchanan M.E. Federici, and S.M. Seiler, J. Med. Chem., 1993 36, 3871-3883.
9. R. Dahiya, D. Pathak and S. Bhatt, J. Saudi. Chem. Soc., 2006, 10, 1, 165-176.
10. A.W. Bauer, W.M. Kirby and M. Turck, Amer. J. Clin. Path., 1966, 45, 493-496.
11. L.C. Garg and C.K Atal, Indian J. Pharm. Sci., 1963, 59, 240-245.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





