IJCRR - 1(2), November, 2009
Pages: 12-22
Print Article
Download XML Download PDF
SYNTHESIS AND BIOLOGICAL SCREENING OF CYCLIC HEPTAPEPTIDE
Author: Komalpreet KaurRamninder Kaur
Category: Healthcare
Abstract:A new bioactive cyclic heptapeptide cyclo(Gly-Tyr-Val-Pro-Leu-Trp-Pro) was synthesized using the solution phase technique by cyclization of the linear peptide Boc- Gly-Tyr-Val-Pro-Leu-Trp-Pro after proper deprotection at carboxyl and amino terminals. All the coupling reactions were performed at room temperature utilizing dicyclohexylcarbodiimide (DCC) as the coupling agent and N-methylmorpholine (NMM) as the base. Structures of all new compounds were characterized by IR and 1HNMR. The synthesized cyclopeptide was
screened for antimicrobial and anthelminitic activities and found to exhibit good antibacterial activity against Bacillus subtilis and moderate antifungal activity against Candida albicans and Asperigillus niger. In
addition the cyclic peptide was found to exhibit good anthelminitic activity against earthworms Eudrilus species.
Keywords: cyclic heptapeptide, antimicrobial activity, anthelminitic activity.
Full Text:
Cyclic congeners possess unusual or modified amino acid residues and exhibit there bioactivities through binding to corresponding enzyme. This characteristic feature can allow bioactive cyclopeptides to act as therapeutic agents in this resistant world. Cyclopepetides having multiple peptide bonds are concerned with a wide spectrum of biological activities such as antimicrobial, anti-inflammatory, antimalarial, cyctotoxic, and antifungal activities. Cyclic peptides are more important compounds for medicinal purposes and represent an important class of natural products. Since only minute quantities are obtained from natural resources many of these compounds were attempted to synthesize in the laboratory. Keeping in view the biological potential of cyclic peptide as well as to obtain a bioactive compound in a good yield, the present investigation aimed at synthesis of cyclic heptapeptide cyclo(Gly-Tyr-Val-Pro-Leu-Trp-Pro) in a convenient and economical manner. Synthesized cyclic heptapeptide was evaluated for pharmacological activities. The antibacterial and antifungal activities were carried out against variety of pathogenic microorganism like Escherichia coli, Staphylococcus aureus, Bacillus subtilis and Candida albicans, Asperigillus niger. The anthelminitic activity was carried out against Eudrilus species of earthworms.
MATERIALS AND METHODS
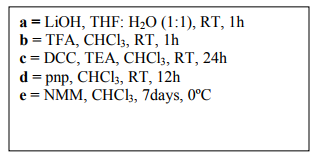
Melting points were determined and uncorrected. The amino acids, di-tert-butyl pyrocarbonate (Boc2O), p-nitrophenol (pnp), DCC and NMM were obtained from Spectrochem Limited and Sd-fine-chem Limited, Mumbai, India. The IR spectra were recorded on a Perkin Elmer Fourier transform infrared spectrophotometer using KBr pellets. The 1HNMR spectra were recorded on the Bruker Avance II-400 NMR spectrometer using CDCl3 as the solvent. The purity of all the compounds was controlled by TLC on silica gel G plates. Chloroform:Methanol (9:1 v/v) was used as developing solvent system and dark brown spots were detected on exposure to iodine vapours in a tightly closed chamber. The physical data of synthesized compounds is listed in Table 1. The scheme of synthesis is given in Scheme 1.
Synthesis of Boc amino acids (1-3) :
L-tyrosine (1.81gm, 10mmol) was dissolved in 10 ml of sodium hydroxide (1 mol L-1 ) and 10 ml of i-propanol. ditert.butylpyrocarbonate (3 ml, 13 mmol) in 5 ml of i-propanol was added followed by 10 ml of sodium hydroxide (1 mol L-1 ) to the resulting solution. The solution was stirred at room temperature for 2 hr, washed with 10 ml of light petroleum ether (b.p. 40-60 C), acidified to pH 3.0 with 1 mol L-1 sulphuric acid and finally extracted with chloroform (3 x 20 ml). The organic layer was dried over anhydrous sodium sulphate and evaporated under reduced pressure to give crude product. The crude product was purified by recrystallization from methanol and ether at 0 C to get pure Boc-Tyrosine (1). Similarly, Boc-leucine (2) and Boc-proline (3) were prepared by stirring ditert.butylpyrocarbonate (3 ml, 13 mmol) with Boc-proline (1.15 gm, 10 mmol) and Boc-leucine (1.31gm, 10 mmol) respectively.
Synthesis of L-amino acid methyl ester hydrochlorides (4-7) :
Thionyl chloride (0.73mL, 10 mmol) was slowly added to methanol (50 mL) at 0 C and 1.15gm of L- proline (10 mmol) was added to the above solution. The resulting mixture was refluxed for 9 hrs at 110 C. Methanol was evaporated and the residue was triturated with ether at 0 C until excess dimethyl sulphite was removed. The crude product was purified by recrystallization from methanol and ether at 0 C to get L-proline methyl ester hydrochloride (4). Similarly, L-valine methyl ester hydrochloride (5), L-tryptophan methyl ester hydrochloride (6) and glycine methyl ester hydrochloride (7) were prepared by refluxing 1.17 gm of L-valine (10 mmol), 2.04 gm of L-tryptophan (10 mmol) and 0.75 gm of glycine (10 mmol) with 50 ml methanol in the presence of 0.73 ml of thionyl chloride (10 mmol).
Synthesis of Boc-dipeptide methyl esters (8-10):
To a mixture of 1.67gm compound 5 (10 mmol) in 20 ml of chloroform, 2.3 ml of N- methylmorpholine (21mmol) was added at 0 C. The reaction mixture was stirred for 15 min. 2.81gm compound 1 (10mmol) in 20 ml chloroform and 2.1gm of DCC (10mmol) were added under stirring to the above mixture. After 36 hrs, the reaction mixture was filtered and the residue was washed with 30 ml of chloroform and added to the filterate. The filterate was washed with 5% sodium hydrogen carbonate and saturated sodium chloride solution (25 ml each). The organic layer was dried over anhydrous sodium sulphate, filtered and evaporated in vacuum. The crude product was recrystallized from mixture of chloroform and petroleum ether (b.p. 40-60 C) followed by cooling at 0 C to get Boc-Tyr-Val-OMe (8). Similarly Boc-Leu-Trp-OMe (9) and Boc-Pro-Gly-OMe (10) were prepared by stirring compounds 2 and 3 with amino acid methyl ester hydrochlorides 6 and 7, respectively in the presence of DCC and NMM.
Deprotection of dipeptides at carboxyl end (8a, 9a) :
To a solution of 3.94 gm of compound 8 (10 mmol) in 36 ml of THF/H2O (1:1), 0.36 gm lithium hydroxide (15mmol) was added at 0 C. The mixture was stirred at room temperature for 1 hr, and acidified to 1 pH 3.5 with 0.5 mol L-H2SO4. The aqueous layer was extracted with diethyl ether (3 x 25 ml). Combined organic extracts were dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was recrystallized from methanol and ether to get Boc-Tyr-Val-OH (8a). Similarly compound 9 was hydrolyzed under alkaline conditions to obtain Boc-LeuTrp-OH (9a).
Deprotection of dipeptide at amino end (10a):
Compound 10 (2.86 gm, 10mmol) was dissolved in 15 ml of chloroform and treated with 2.28 gm of trifluoroacetic acid (20 mmol). The resulting solution was stirred at room temperature for 1 hr and washed with 25 ml of saturated sodium hydrogen carbonate solution. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by recrystallization from mixture of chloroform and light petroleum ether (b.p. 40-60 C) to get pure Pro-Gly-OMe (10a)
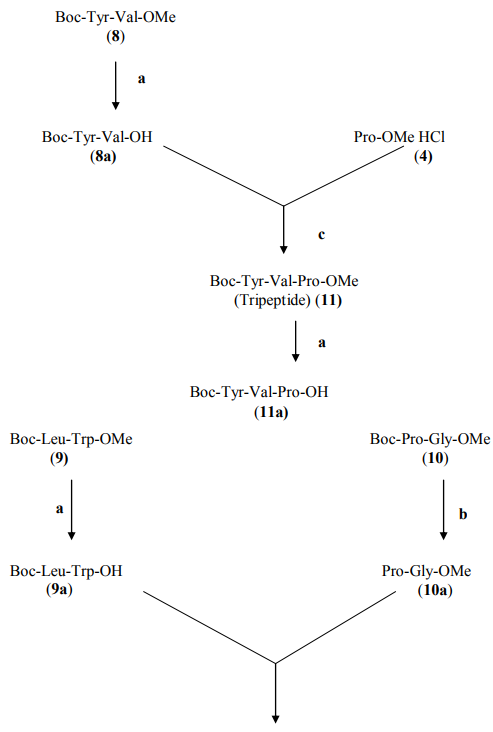
Synthesis of Boc-tri/tetrapeptide methyl esters (11, 12) :
To synthesize Boc-Tyr-Val-Pro-OMe (11), 3.80 gm of dipeptide unit 8a (10 mmol) was coupled with 1.66 gm of amino acid methyl ester hydrochloride 4 (10 mmol) in the presence of DCC and NMM following the same procedure as adopted for the synthesis of Boc-dipeptide methyl esters 8- 10. Similarly Boc-Leu-Trp-Pro-Gly-OMe (12) was prepared by coupling 3.36 gm of deprotected dipeptide unit 9a and 1.86 gm of 10a using DCC as the coupling agent and NMM as the base.
Synthesis of Boc-heptapeptide methyl ester (13):
To synthesize Boc-Tyr-Val-Pro-LeuTrp-Pro-Gly-OMe (13), 4.77 gm of tripeptide unit 11 mmol was deprotected at carboxyl end to get Boc-Tyr-Val-Pro-OH (11a) following the same procedure as adopted for the synthesis of compounds 8a and 9a from compounds 9 and 10, respectively. Tetrapeptide unit (12) (5.85 gm, 10 mmol) was deprotected at amino end to get Leu-Trp-Pro-Gly-OMe (12a) following the same procedure as adopted for the synthesis of compounds 10a from compound 10. The deprotected tripeptide unit 11a (4.77 gm, 10 mmol) and 4.85 gm of tetrapeptide unit (10 mmol) were coupled in the presence of DCC and NMM to get linear heptapeptide unit 13 under the same experimental conditions as adopted for the synthesis of Boc-dipeptide methyl esters (8- 10).
Synthesis of cyclic heptapeptide, cyclo(Gly-Tyr-Val-Pro-Leu-Trp-Pro) (14):
To synthesize cyclo(Gly-Tyr-Val-ProLeu-Trp-Pro) (14), 9.44 gm of linear heptapeptide unit(10 mmol) (13) was deprotected at carboxyl end using 0.36 gm of LiOH (15 mmol) to get Boc-Tyr-Val-ProLeu-Trp-Pro-Gly-OH (13a) following the same procedure as adopted for the synthesis of compounds 8a and 9a from compounds 8 and 9 respectively. The deprotected heptapeptide unit 13a (4.65 gm, 5 mmol) was dissolved in 50 ml of CHCl3 at 0 C. To the above solution, 0.94 gm of pnp (6.7 mmol) was added and stirred at room temperature for 12 hrs. The reaction mixture was filtered and the filtrate was washed with 10% NaHCO3 solution (3 x 15 ml) until excess of pnp was removed and finally washed with 5% HCl (2 x 10 ml) to get the corresponding p-nitrophenyl ester Boc-Tyr-Val-Pro-Leu-Trp-Pro-Gly-O-pnp (13b). To compound 13b (4.20 gm, 4 mmol) dissolved in 35 ml of CHCl3, 0.91 gm of TFA (8 mmol) was added, stirred at room temperature for 1 hr and washed with 10% NaHCO3 solution (2 x 25 ml). The organic layer was dried over anhydrous sodium sulphate to get Tyr-Val-Pro-Leu-Trp-ProGly-O-pnp (13c), which was dissolved in 25 ml of CHCl3 and 2.3 ml of NMM (21 mmol) was added. Then all the contents were kept at 0 C for 7 days. The reaction mixture was washed with 10% NaHCO3 until the byproduct p-nitrophenol was removed completely and finally washed with 5% HCl (3 x 15 ml). The organic layer was dried over anhydrous sodium sulphate. Finally, chloroform was distilled off and the crude cyclized product was crystallized from CHCl3 and n-hexane to get the pure compound 14.
Antibacterial Activity:
The synthesized cyclic peptide was screened for antibacterial activity against Escherichia coli, Staphylococcus aureus and Bacillus subtilis using modified Kirby-Bauer disc diffusion method. A spore suspension in sterile distilled water was prepared from five days old culture of the test bacteria growing on nutrient broth media. About 20 ml of the growth was transferred into sterilized petri plates and inoculated with 1.5 ml of the spore suspension. Each petri plate was divided into five equal portions along the diameter to place one disc. Three discs of test sample were placed on three portions together with one disc reference drug ciprofloxacin (50pg/ml) and a disc impregnated with the solvent DMF as negative control. The test samples were tested at the concentrations 25, 50, 100 pg/ml. The petri plates inoculated with bacterial cultures were incubated at 37 C for 18 hrs. Diameters of the zone of inhibition were calculated in triplicate sets. The diameters obtained for the test sample were compared with that produced by the standard drug ciprofloxacin. The results are shown in Table 2.
Antifungal Activity:
The synthesized cyclic peptide was screened for antifungal activity against Candida albicans and Asperigillus niger. A spore suspension in normal saline was prepared from culture of the test fungi on sabouraud?s broth media. After transferring growth media, petri plates were inoculated with spore suspension. After drying, wells were made using agar punch and the test samples, reference drug (griseofulvin) (50pg/ml) and negative control (DMSO) were placed in labeled wells in each petri plate. The test samples were tested at the concentrations 25, 50, 100 µg/ml. The petri plates inoculated with fungal cultures were incubated at 25 C for 48 hrs. Diameters of the zone of inhibition were calculated in triplicate sets. The diameters obtained for the test sample were compared with that produced by the standard drug griseofulvin. The results are shown in Table 2.
Anthelmintic Activity:
The anthelminitic activity was carried out against earthworms Eudrilus species by Garg and Atal method at 2 mg/ ml concentration. Suspension of samples was prepared by triturating synthesized cyclic peptide (200 mg) with Tween 80 (0.5 %) and distilled water. The resulting mixture was stirred using mechanical stirrer for 30 min. The suspensions were diluted to contain 0.4 % w/v of the test samples. Suspension of the standard drug albendazole was prepared with the same concentration in a similar way. Three sets of five earthworms of almost similar sizes were placed in petri plates containing 50 ml suspension of Tween 80 (0.5 %) and distilled water. The paralyzing and death times were noted and their mean was calculated for triplicate sets. The death time was ascertained by placing the earthworms in warm water (50 C) which stimulated the movement. The results were shown in Table 3.
RESULTS AND DISCUSSION
The results revealed that newly synthesized cyclic peptide 14 at 50 pg ml-1 concentration exhibited highest zone of inhibition against B. subtilis. The cyclic peptide 14 at 50 pg ml-1 concentration exhibited moderate antifungal activity against A. niger and C. albicans. Anthelmintic activity data revealed that cyclic peptide 14 possessed moderate anthelmintic activity against Eudrilus sp. comparable to that of reference drug. Cyclization of linear heptapeptide fragment 13 was indicated by disappearance of absorption bands at 1736 cm-1 (C=O stretching of ester) and 1392, 1367 cm-1 (CH band of t butyl group) and presence of additional Amide I and Amide II bands at 1659 and 1539 cm-1 in IR spectrum of synthesized cyclic peptide 14. Formation of cyclic peptide was further confirmed by disappearance of singlets at 3.62 and 1.60 ppm corresponding to three protons of methyl ester group and nine protons of t butyl group of di-tert.butylpyrocarbonate in 1HNMR spectrum of the cyclic heptapeptide 14 showed characteristic peaks confirming the presence of all seven amino acid moieties.
ACKNOWLEDGEMENTS
The authors are thankful to the Head, Department of Chemistry for providing research facilities, SAIF Department, Punjab University, Chandigarh (India), for providing spectral details in time.
TABLE 1 : PHYSICAL DATA OF THE SYNTHESIZED COMPOUNDS
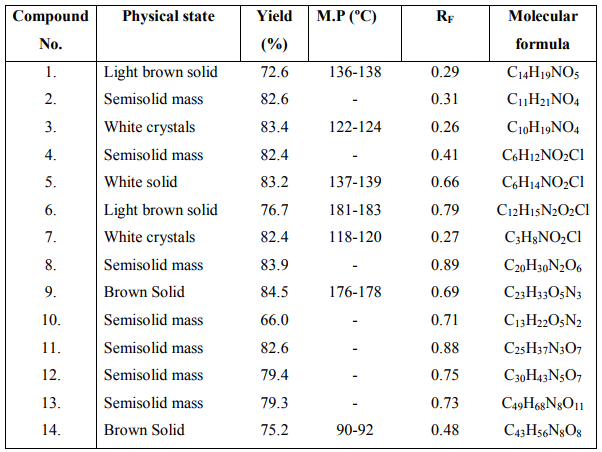
Table 2 : ANTIBACTERIAL AND ANTIFUNGAL ACTIVITY OF COMPOUND 14:
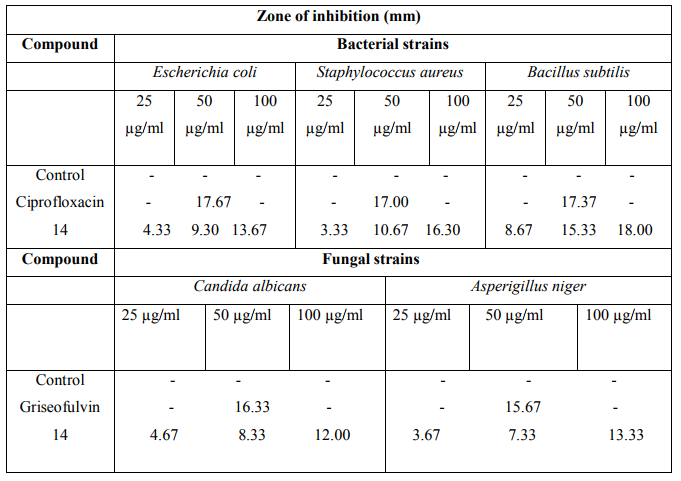
TABLE 3 : ANTHELMINTIC ACTIVITY OF COMPOUND14 :
TABLE 4: SPECTRAL DATA OF SYNTHESIZED COMPOUNDS

Scheme (1)
Synthesis of Cyclo(glycyl-tyrosinyl-valyl-prolyl-leucyl-tryptophanyl-prolyl)

References:
1. Poojary B. and Belagali S.L. Synthetic studies on cyclic octapeptides, Yunnanin F and Hymenistatin. Eur. J. Med. Chem., 2005; 40 : 407-412.
2. Dahiya R. and Pathak D. Synthetic studies on a natural cyclic tetrapeptide Halolitoralin C. J. Pharm. Res., 2006; 5, 3 : 69-73.
3. Morita H., Yun Y.S., Takeya K., Itokawa H. and Shirota O., A cyclic heptapeptide from Vaccaria Segetalis. Phytochemistry, 1996; 42, 2 : 439-441.
4. Dahiya R., Pathak D., Himaja M. and Bhatt S. First total synthesis and biological screening of hymenamide E. Acta Pharm., 2006; 56 : 399-415.
5. Bauer A.W., Kirby W.M. and Turck M. Amer. J. Clin. Path., 1966; 45 : 493-496.
6. Garg L.C. and Atal C.K. Indian J. Pharm. Sci., 1963; 59 : 240- 245.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





