IJCRR - 3(3), March, 2011
Pages: 73-80
Print Article
Download XML Download PDF
PRODRUGS: AN APPROACH TOWARDS BETTER TARGETTING
Author: Mayank Yadav, Himani Bajaj, Vinod Singh, Mamta Singh
Category: Healthcare
Abstract:Prodrugs, the pharmacologically inactive derivatives of active drugs, are designed to maximize
the amount of active drug that reaches its site of action, through manipulation of the
physicochemical, biopharmaceutical or pharmacokinetic properties of the drug. But new
developments are increasingly taking the concept beyond issues of availability to include
targeting. The development of prodrugs promises to be very effective method for treatment of
diseases in future. This approach has several advantages over conventional drug administration.
In this mini review, prodrugs are discussed with a focus on the viability of the prodrug approach
as a means of attaining targeted drug delivery. Next, several examples of where the prodrug
approach has been used to achieve targeted delivery will be discussed.
Keywords: prodrugs, targeting, targeted drug delivery.
Full Text:
INTRODUCTION
A drug can be defined as a chemical used for treating, curing or preventing disease in human beings or in animals. In the process of treatment, drugs are also used for medical diagnosis and for restoring, correcting, or modifying physiological functions. Conventional drugs suffer from many drawbacks in their performance like site specificity, permeability and resistance. Almost all drugs possess some undesirable physicochemical and biological properties. Their therapeutic efficacy can be improved by minimizing or eliminating the undesirable properties while retaining the desirable ones 1 . This can be achieved through following means. • The biological approach is to alter the route of administration which may or may not be acceptable to patient. • The physical approach is to modify the design of dosage form such as controlled drug delivery of drug. • The third and best approach in enhancing drug selectivity while minimizing toxicity is the chemical approach for design of prodrugs 2 .
The Prodrug Concept
Albert and his coworkers were the first ones to suggest the concept of prodrug approach for increasing the efficiency of drugs in 1950. They described prodrugs as pharmacologically inactive chemical derivatives that could be used to alter the physicochemical properties of drugs, in a temporary manner, to increase their usefulness and/or to decrease associated toxicity2 .Subsequently such drug-derivatives have also been called „latentiated drugs?, „bioreversible derivatives?, and „congeners?, but „prodrug? is now the most commonly accepted term. Thus, prodrug can be defined as a drug derivative that undergoes biotransformation enzymatically or nonenzymatically, inside the body before exhibiting its therapeutic effect. According to IUPAC (International Union of pure and applied chemistry): Prodrug is defined as any compound that undergoes biotransformation before exhibiting its pharmacological effects 3 .
Characteristics of a Prodrug In recent years numerous prodrugs have been designed and developed to overcome barriers to drug utilization such as
4 : ? Low oral absorption properties
? Lack of site specificity
? Chemical instability
? Toxicity ? Bad taste
? Bad odour
? Pain at application site 4, 5
Ideal properties of prodrug It should have intrinsic pharmacological activity that means it should not change receptor configuration that is necessary for pharmacological response. It should rapidly transform into the active from where desired and metabolic fragment, apart from the active drug should be nontoxic 6 .
Limitation of prodrug 7 The problem associated with prodrug design is its toxicity which is due to Formation of unexpected metabolite from the total drug conjugates. Toxicity may be due to inert carrier generated by cleavage of promoiety and drug conjugate which is converted into toxic metabolite. The prodrug might consume a vital cell constituent such as glutathione during its activation stage which causes depletion of prodrug 8 .
Classification of Prodrugs A)
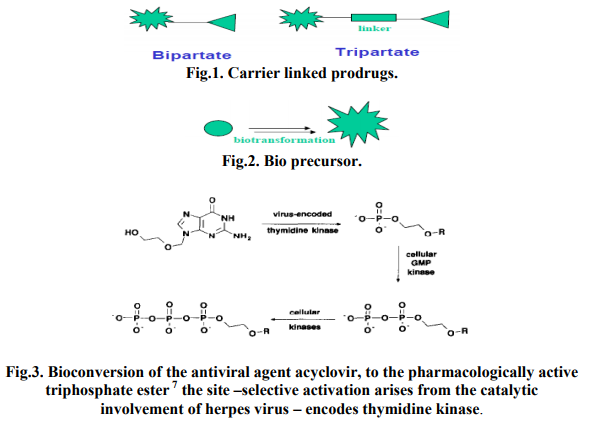
Carrier linked prodrug Contain a group that can be easily removed enzymatically (such as ester) to reveal the true drugs as shown in fig.1.Ideally the group removed is pharmacologically inactive and nontoxic while the connecting bond must be labile for efficient activation in vivo. Prodrugs are the ones where the active drug is covalently linked to an inert carrier transport moiety. They are generally esters or amides. Such prodrugs have greatly modified lipophilicity due to the attached carrier and the active drug is released by hydrolytic cleavage, either chemically or enzymically 9 . It can be further subdivided intoBipartate- Composed of one carrier (group) attached to the drugs. Tripartat- Carrier group is attached via linker to drug. Mutual Prodrugs- Two drugs linked together.
B) Bio precursors Metabolized into a new compound that may itself be active or further metabolized to an active metabolite as shown in fig.2 (e.g. amine to aldehyde to carboxylic acid) 10
PRODRUG MEDIATED TARGETED DELIVERY
Targeting viruses
The topic of targeted delivery of antiviral agents has been recently reviewed 11. One of the best and simplest examples of targeted drug delivery via a prodrug approach is the antiviral agent. Acyclovir (9-(2- hydroxyethoxymethyl) - guanine). As shown in Fig. 3, its targeting is achieved primarily by site-selective activation. The herpes virus encoded enzyme, pyrimidine deoxynucleoside (thymidine) kinase, is responsible for converting acyclovir to its phosphate monoester. Subsequently, cellular enzymes catalyse the conversion of the monoester to the di- then triphosphorylated species. This latter species is the pharmacologically active one, and the enzymatic conversion to triester occurs to a significantly greater extent in the herpesinfected cells. Because of its site-specific activation, acyclovir displays a high therapeutic activity against herpes virus, essentially no activity against adenovirus, minimal metabolic degradation following systemic administration, and very low toxicity against uninfected host cells 12, 13.
Targeting the colon
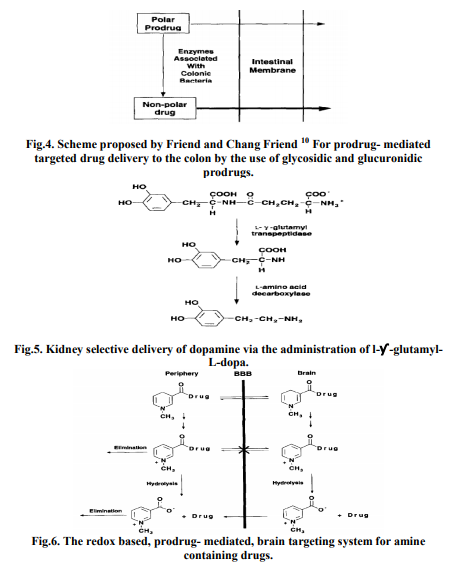
A fundamentally simple example of targeting which exploits both site-specific transport and activation is the colon-specific delivery of drugs illustrated in Fig. 4. With this approach, prodrugs are formed by coupling the drug to a hydro- philizing promoiety that is susceptible to cleavage by enzymes secreted by the bacterial microflora associated with the lower gastrointestinal tract 14 . Following oral administration of the prodrug, drug absorption in the stomach and small intestines is decreased due to the polar nature of the promoiety; therefore, greater levels of the drug, in the form of the prodrug, can reach the colon. Within the colon, the bacterially derived enzymes catalyze the conversion of the prodrug to the more lipophilic drug which is now available for absorption through the colonic membrane. This targeting concept has been recently reviewed Glycosidic and glucuronidic prodrugs, of agents such as dexamethasone, naloxone, and menthol, that exploit bacterial glycosidases and glucuronidases have been studied for their colon-targeting potential 15 .
Targeting the kidney
Site-selective prodrug activation, by exploiting the relatively high response site activity of an enzyme, has also been achieved with targeted delivery to the kidney. For example, dopamine was found to selectively accumulate in the kidney following the intraperitoneal administration of the double prodrug, y-glutamyl-L-dopa, to mice 16. As shown in Fig. 5, the prodrug is activated by the sequential catalytic actions of two enzymes that possess high activity in the kidney. First, y-glutamyl transpeptidase catalyzes the cleavage of the y-glutamyl linkage: the L-dopa which is formed is then decarboxylated to dopamine by L-amino acid decarboxylase. The end result is that dopamine is more readily available to exert its therapeutic effect (i.e., renal vasodilation) at the response site while causing less effects at non-response sites (as was demonstrated by an unchanged systemic blood pressure).
Targeting the liver
To achieve targeted drug delivery to the liver, researchers have attempted to exploit site-selective transport pathways. One such pathway is the bile acid transport system associated with the sinusoidal membrane of hepatocytes 17. The bile acid prodrugs of several compounds, such as chlorambucil, thyroid hormone (L-T). and HMG-CoA reductase inhibitors 18 have demonstrated some degree of hepatic targeting.
Targeting the brain
To preferentially deliver amine-containing drugs to the brain 19 have developed a prodrug delivery system which exploits the oxidative conversion of a dihydropyridine promoiety to its corresponding pyridinium salt (Fig. 6). With this approach, an aminecontaining drug is coupled to a lipohilic dihydropyridine promoiety that facilitates the penetration of the drug, in the form of the prodrug, through the blood-brain barrier (BBB). Oxidation of the dihydropyridine functionality in the peripheral compartments results in the formation of the corresponding polar pyridinium salt which can be readily excreted from the body, whereas formation of the pyridinium salt in the CNS results in site retention because of the poor permeation characteristics of the charged species and the permeability characteristics of the BBB. Subsequent cleavage of the pyridinium salt promoiety releases the drug which is now present at elevated levels within the brain. The proof-of concept has been has been demonstrated for a wide variety of aminecontaining drugs.
Targeting with antibodies
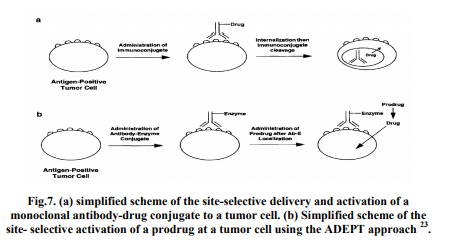
Selective activation of a prodrug of an anticancer agent at tumor sites is severely limited by the commonality of enzymes associated with normal and neoplastic tissues. Therefore, two approaches utilizing monoclonal antibodies20 have been studied as a way of selectively activating a prodrug at the tumor site. These approaches, which have received considerable attention over the last decade, are Antibody- Drug Conjugates and Antibody-Directed Enzyme Prodrug Therapy (ADEPT). Antibody-drug conjugates Antibody-drug conjugates (immune conjugates) are macromolecular prodrugs that are formed by covalently linking cytotoxic agents to monoclonal antibodies reactive with tumor associated antigens. A number of chemical coupling methods have been utilized to produce the drug-antibody conjugates, and several tumor-associated antigens have been identified, and the respective monoclonal antibodies have been produced. In order for targeting to be achieved, the drug must be cleaved from the antibody after the immune conjugate binds to a tumor cell; this usually occurs intracellularly (e.g. lysosomal degradation) after internalization of the conjugate Fig. 7a shows a simplified depiction of this process 21 .
Antibody-directed enzyme prodrug therapy (ADEPT)
A simplified scheme of the ADEPT approach to targeted drug delivery is shown in Fig. 7b. Ideally, administration of an enzyme, which is covalently linked to a monoclonal antibody, binds selectively to the respective tumor-associated antigen. After the antibody-enzyme conjugate (Ab-E) has localized within the tumor and has been cleared from non-target sites, a prodrug, that is a substrate for the enzyme, is administered. Upon contact with the targeted enzyme, the prodrug is converted to the drug at the tumor site. This targeting approach has been well reviewed. An important dosing consideration with the ADEPT approach is optimizing the time interval between administration of the Ab-E and the prodrug. To attain adequate tumor uptake, high plasma and extracellular fluid levels of the Ab-E should typically be maintained for several hours 22, 23. Once adequate tumor levels are achieved (and prior to prodrug administration), sufficient time should be allowed for significant plasma clearance or inactivation of the non-tumor-associated AbE to minimize prodrug activation at non tumor sites (a potential source of toxicity).
CONCLUSION
The area of prodrug-mediated targeted drug delivery has made many strides within the last decade; however, there is still a great need for additional work to further improve existing approaches and to develop newer ones. At present prodrug are not prevalent in clinical use, in future there will be prodrugs for every known drug to make them effective in treatment. Drug discovery and prodrug development appear to be complementary for the generation of target specific medicines of future. The sole use of a prodrug to achieve targeted drug delivery is limited unless the target site possesses a unique enzyme system for activating the prodrug (as in the case of acyclovir). Hence, the combination of a prodrug approach with an additional approach which facilitates the targeting, as exemplified by the ADEPT. At present the research in this area is at a nascent stage due to lack of information, regarding all enzymes or receptors, most suitable for targeting purposes. As the unrevealing of the microbiological details of affected targets become clear, prodrug development will surely decrease side/toxic effect of drugs and also trigger development of more potent primary drugs.
References:
1. P K Banerjee and G L Amidon, Design of Prodrugs, (Ed.) H Bundgaard, New York: Elsevier; pp. 93-133, 1985. F M H de Groot, E W P Damen and H W Scheeren, Current Medicinal Chemistry 8, p.1093, 2001
. 2. Tripathi K.D. Essentials of medical pharmacology.4th edition , 25
3. IUPAC Pure and appl. chem., 1998, 70, 1129-1143.
4. Mayer D.L., Jungheim L.N, Law K.L., Mikolajczyle S.D.,Shapherd T.A., Cancer Res.,1993, sep.1:53(17):3956-63.
5. S.Kenneth, Prodrug topical and ocular drug delivery, 17- 22.
6. Kawakami S.,Yamishita.F., Hashida M., Adv. Drug Delivery Review,Vol 45,Issue 1, 6 dec 2000,77-88.
7. Meijer, D.K.F., Jansen, R.W. and Molema, G. (1992) Drug targeting systems for antiviral agents: options and limitations. Antiviral Res. 18, 215-258.
8. Wilding. I.R., Davis, S.S. and O?Hagan, D.T. (1994) Targeting of drugs and vaccines to the gut. Pharmacol. Ther. 62. 97-124.
9. Tozer, T.N.. Friend, D.R. and McLeod, A.D. (1995) Kinetic perspectives on colonic delivery. STP Pharm. Sci. 5, S- 12.
10. Friend, D.R. and Chang, G.W. (1985) Drug glycosides: potential prodrugs for colon-specific drug delivery. J.Med. Chem. 28, 51-57.
11. Simpkins, J.W., Smulkowski, M., Dixon, R. and Tuttle. R.E. (1988) Evidence for the delivery of narcotic antagonists to the colon as their glucuronide conjugates. J. Pharmacol. Exp. Ther. 244, 195-205.
12. Haeberlin. B., Rubas, W., Nolen, H.W. and Friend. D.R. (1993) In vitro evaluation of dexamethasone-P-oglucuronide for colon-specific drug delivery. Pharm. Res. 10 1553-1562.
13. Anwer, M.S., Kroker, R. and Hegner. D. (1976) Cholic acid uptake into isolated rat hepatocytes. Hoppe- Seyler?s Z. Physiol. Chem. 357 1477-1486.
14. Stephan, Z.F., Yurachek, E.C., Sharif, R., Wasvary. J.M.. Steele, R.E. and Howes, 80 International Journal of Current Research and Review www.ijcrr.com Vol. 03 issue 03 Mar 2011 C. (1992) Reduction of cardiovascular and thyroxine-suppressing activities of LT, by liver targeting with cholic acid. Biochem. Pharmacol. 43. 1969-1974.
15. Kramer, W., Wess, G.. Enhsen, A., Bock, K., Falk, E., Hoffman, A., Neckermann, G., Gantz, D., Schulz, S., Nickau, L.. Petzinger, E., Turley, S. and Dietschy, J.M. (1994) Bile acid derived HMG-CoA reductase inhibitors. Biochim. Biophys. Acta 1227, 137-154.
16. Neufeld, E. and Ashwell, G. (1979) Carbohydrate recognition systems for receptor-mediated pinocytosis. In: W. Lennarz (Ed.), Biochemistry of Glycoproteins and Proteoglycans, Plenum Press, New York, pp. 241-266.
17. Bodor and co-workers Bodor, N., Farag. H.H. and Brewster, M.E. (1981) Sitespecific, sustained release of drug to the brain. Science 214, 1370-1372.
18. Pop, E., Brewster, M.E. and Bodor, N. (1991) Sitespecific delivery of the central nervous system acting amines. Drugs Future 16, 919-944.
19. Kohler, H. and Milstein. C. (1975) Continuous cultures of fused cells secreting antibodies of predefined specificity. Nature 256, 495. Waldmann, T.A. (1991) Monoclonal antibodies in diagnosis and therapy. Science 252, 1657-1662
20. Garnett, M.C., Embleton. M.J., Jacobs, E. and Baldwin, R.W. (1985) Studies on the mechanism of action of an anti-bodytargeted drug-carrier conjugate. AntiCancer Drug Des. 1. 3-12.
21. Deonarain, M.P. and Epenetos, A.A. (1994) Targeting enzymes for cancer therapy: old enzymes in new roles. Br. J. Cancer 70, 786-794.
22. Bagshawe, K.D., Sharma. SK., Springer, C.J. and Rogers. G.T. (1994) Antibody directed enzyme prodrug therapy (ADEPT). Ann. Oncol. 5, 879-891
. 23. Wallace, P.M. and Senter, P.D. (1994) Selective activation of anticancer prodrugs by monoclonal antibodyenzyme conjugates. Methods Find. Exp. Clin. Pharmacol. 16. 505-512.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





