IJCRR - 3(3), March, 2011
Pages: 42-51
Print Article
Download XML Download PDF
IPOPROTEIN LIPASE GENE POLYMORPHISM AT Pvu II RESTRICTION SITE AND CORONARY ARTERY DISEASE
Author: A. Cholarajan, A.Sharmila Princy
Category: Healthcare
Abstract:The present study was made to find the PvuII restriction enzyme polymorphism of lipoprotein lipase
(LPL) gene and its association with angiographically defined coronary artery disease (CAD). Lipoprotein
lipase (LPL) plays a central role in lipid metabolism by hydrolyzing triglyceride in chylomicrons and
VLDL. Polymorphic variants of the LPL gene are common and might affect risk of CAD.Two milliliter
of blood samples were collected from 30 patients, and 30 control subjects. Genomic DNA was isolated
from the blood samples and subjected to PCR-RFLP analysis for determination of the genotype with
regard to the Pvu II polymorphism of LPL gene. The amplified product was digested with restriction
enzyme Pvu II and genotyped by through electrophoresis in 2.5% agarose gel. For the PvuII genotypes,
within the CAD group(n=30), the +/- genotype (+ presence of Pvu II restriction site, - absence of Pvu II
restriction site) was found in 12 individuals (40%),whereas 10(33%) carried the +/+,and 8(27%) carried
the -/- genotype. In the control group (n=30), the -/- genotype was found in 6 subjects (20%), 13 (43%)
carried the +/+ genotype and 11(37%) carried the +/- genotype. The genotype frequency distribution was
significantly different in the CAD and control study group. The most frequent genotype among CAD
patients was (-/-), this genotype was more frequent in patients than in control subjects.There was a
difference in the distribution of LPL-PvuII genotypes between the healthy subjects and the patients with
CAD. The common LPL polymorphic allele, the PvuII (+) or (-) is modestly associated with CAD.
Genetic variants of LPL deserve further evaluation as risk factors for CAD.
Keywords: Lipoprotien lipase; coronary artery disease ; pvu II restriction site ; Enzyme
Full Text:
INTRODUCTION
Coronary Artery Disease (CAD) is the most common form of heart disease. This condition occurs when the coronary arteries, that supply oxygen- rich blood to the heart muscle, gradually become narrowed or blocked by plaque deposits. The plaque deposits decrease the space through which blood can flow. Poor blood flow can „starve? the heart muscle and lead to chest pain. A heart attack results when blood flow is completely blocked, usually by a blood clot forming over a plaque that has ruptured (Lusis et al., 2000). Lipoprotein lipase (LPL;triacylglycerolprotein acyl hydrolase, EC 3.1.1.34)catalyzes the hydrolysis of core triacylglycerol of circulating very low density lipoprotein(VLDL) and chylomicrons generating fatty acids for storage in adipose tissue or oxidation in muscle. This enzyme therefore plays a crucial role in plasma lipoprotein processing and energy metabolism in general (Eckel,1987).LPL is a glycoprotein synthesized and secreated by a variety of parenchymal cells, including adipocytes,skeletal and cardiac muscle cells and macrophages. After secreation it becomes bound to glycosaminoglycans on the luminal surface of capillaries. Apolipoprotein CII, which is present on the surface of VLDL and chylomicrons, was shown to act as a cofactor for LPL (Olivecrona et al., 1997).
The importance of LPL in the regulation of overall lipid metabolism and transport in humans and other animals is well documented (Cryer, 1981).LPL is expressed by the parenchymal cells of extra hepatic tissue and catalyses the hydrolysis of the triacylglycerol componenet of chylomicrons and very low density lipoprotein (VLDL),thereby providing non-esterified fatty acids and 2-monoacylglycerol for tissue utilization .Because the circulating lipoproteins are in general, too large to cross the vascular endothelial cells, to which the enzyme is attached via highly charged membrane bound chains of heparin sulfate proteoglycans (HSPG) (Godota et al, 1992) .Although adipose tissue and muscle parenchymal cells are the major source of LPL synthesis, from where the mature enzyme is secreted by macrophages. It is the source of LPL that has been implicated to play a major role in the pathogenesis of atherosclerosis.
LPL gene, mapped to the short arm human chromosome8, contain 10 exons, separated by 9 introns (Deep et al., 1989).Gene analysis showed many mutations involving the noncoding and coding sequences of the human LPL gene (Murthy et al., 1996).Several restriction fragment length polymorphisms (RFLPs) have been identified at the LPL gene. The occurrence of alternate types of nucleotides at the same position in the nucleic acid sequence with no concomitant apparent phenotypic differences is generally referred to as polymorphism and it can be easily detected if it leads to restriction site alterations. The most extensively investigated of these are the PvuII (Fisher et al., 1987) and HindIII (Heinzmann et al., 1987) sites. PvuII polymorphism is the result of change in the restriction site of the LPL gene 6th intron, 1.57kb from SA site (Oka et al., 1989). The region containing PvuII site resembles the splicing in its homology to the consensus sequence required for 3?-splicing and the formation of the lariat structure, suggesting that C497- T(CAG CTG-TAG CTG) change may interfere with correct splicing of mRNA.
DNA polymorphism is a useful marker to analyze disorders with genetic backgrounds, even when the genetic cause of the disease has not been elucidated (Godotaet et al.,1992).A number of DNA polymorphism have been investigated for their possible linkage with hereditary predisposition to common polygenic disorders, such as dyslipidemia (Lusis et al.,1988).Several trails explored association between LPL gene for an association between genotypes identified by the PvuII restriction fragment length polymorphism and plasma triglyceride levels (Chamberlain et al.,1989;Wang et al.,1996),but other failed to find significant association (Heizmann et al.,1991;Jemaa et al.,1995).
MATERIALS AND METHODS
SAMPLE COLLECTION
The blood samples were collected from the patients between the age group of 30-60 from inpatients and outpatient department of Frontier Life Line, Dr.KM.Cherian Heart Foundation, Chennai. Plasma was isolated for subsequent analysis of measuring sugar level, HDL, LDL, Total cholesterol and Triglycerides were carried out in the clinical Biochemistry laboratory of the hospital.
DNA ISOLATION
The genomic DNA was isolated from venous blood by using Phenol chloroform isolation method. The blood samples were obtained in a EDTA (Ethylene diamine tetra acetic acid) coated vacutaines. From this 1ml of the blood was taken into an eppendroff tube and centrifuged at 3000rpm for 30minutes. On centrifugation a white Buffy coat (which has the WBC) was obtained which is removed and transferred to fresh eppendroff tubes. To this, thrice the volume of lysis buffer (0.155M ammonium chloride, 0.1M potassium bicarbonate and 0.1mM EDTA) was added; then Mixed and stored at -20 C for 30minutes which would allow cell lysis to occur. Later the tubes were centrifuged at 2000 rpm for 5 minutes and the supernatant was discarded. To the pellet, 200 l of 0.5M sodium EDTA, 100 l of 10% SDS and 10 l of proteinase k (100 g/ml) were added and left for overnight incubation. The next day to this overnight solution, equal volumes of Tetrahydroxy aminomethyl hydrochloric acid (Tris) saturated phenol (pH 8) was added and mixed well in an overhead shaker for 30minutes. The tubes were centrifuged at 6000 rpm for 10minutes and the upper aqueous layer was removed without disturbing the lower organic layer (containing phenol). To the aqueous layer again obtained equal volumes of Tris saturated phenol, Chloroform and isoamyl alcohol in the ratio 25:24:1 were added and mixed in an overhead shaker for 30minutes. Again the upper aqueous supernatant was taken and further residual phenol was removed by extracting with equal volumes of chloroform and isoamyl alcohol (24:1) and mixed well in an overhead shaker. To the aqueous layer, one tenth the volumes of 3M sodium acetate and equal volumes of chilled ethanol was added and shaken well. The tubes were centrifuged at 12000 rpm for 2 minutes and the supernatant was discarded. The pellet was washed with 70 % ethanol and the DNA obtained was stored in TE (1M Tris, 1mM EDTA) at 4 C.
POLYMERASE CHAIN REACTION Polymerase chain reaction (PCR) is a technique which is designed to permit selective amplification of a specific target DNA sequence or sequences within a heterogeneous collection of DNA sequences. The PvuII genotypes were determined by polymerase chain reaction (PCR) amplification of the polymorphic regions found in intron 6 region of the lipoprotein lipase (Oka et al.,1989), followed by digestion of these amplified fragments with PvuII restriction endonuclease. The PvuII-containing site was amplified using the following primers (Sigma, Bangalore).
Forward 5?- CATCCATTTTCTTCCACAGGG -3?
Reverse 5?-TAGCCCAGAATGCTCACCAGACT- 3?
PCR reaction amplification was carried out in 15 µl reaction volume containing, 1X PCR buffer, 1.5mM MgCl2, 200µM dNTPs, forward and reverse primers 0.5µM respectively, 0.5U Taq DNA polymerase (Sigma, USA ) and Genomic DNA (50ng – 100ng). The thermo cycling procedure was carried out using Thermocycler (MJ Research Inc, USA) for 30 cycles. PCR cycling conditions were 94°C for 5 minutes for initial denaturation followed by 30 cycles of 94 °C denaturation for 45 seconds, 54°C annealing for 30 seconds, 72°C extension for 30 seconds, 72 °C final extension for 3 minutes.
RESTRICTION FRAGMENT LENGTH POLYMORPHISM (RFLP)
The amplified DNA sample was subjected to restriction digestion with 5 units of Pvu II restriction enzyme (Biogene.U.S.A). It was incubated at 37 °C for 3 hours. Then the digested sample was subjected to 2.5% agarose gel electrophoresis.
GEL ELECTROPHORESIS
The gel template was cleaned and its edges were sealed. A solution of 2.5% agarose gel (1X Tris borate EDTA) was prepared and the solution was warmed until the Agarose melted. The melted gel was cooled and 3µl of ethidium bromide was added (10 mg/ml stock). The warm solution was poured into the template and allowed to solidify. The comb and the seal were carefully removed from the solidified gel. The gel was placed in electrophoresis tank and it was filled with 1X TBE buffer. The DNA sample was mixed with loading dye. The sample was loaded carefully with the help of micropipette. The electrode was connected and a voltage of 1-5 volts per cm was applied. The current supply was stopped when the dye had migrated above 70%. The gel was placed carefully on an ultraviolet transilluminator and the DNA bands were viewed. The gel was then photographed and developed.
STATISTICAL ANALYSIS The mean, standard deviation were calculated for each parameter (age, BMI, glucose level, etc.) for both the control and patient samples.
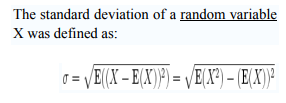
RESULT AND DISCUSSION
The baseline features and biochemical parameters of both control and patients were listed and expressed as mean ± Standard Deviation in the Table-1. The number of genotypes of Pvu II restriction site of LPL gene was computed by gene counting method from the documented gels (Figure 1). The mean age of patients and control subjects was 47.3 + 11.62 and 59.9 + 8.57 years respectively. Among 30 involved patients 25 were male and 5 were female. The patients group presented convincingly lower mean levels of body mass index than control subjects. The mean value of low density lipoprotein level and high density lipoprotein level in patients were lower than the control.


The genotype and allele frequency of the Pvu II restriction site at the LPL locus in patients and control groups were shown in Table-2. The -/- homozygote among the patients and control are 27 and 20% and the +/+ homozygote among the patients and control are 33% and 43 % respectively. Also, the percent of heterozygote +/- varied significantly between the two groups 40% in patients and 37 % in control. The +/- and -/- allele frequency were considerably higher in patients with coronary atherosclerosis than the normal control. The Chi square value in patients and controls were 1.158 and 1.511 respectively, and tested to be present in the Hardy-Weinberg equilibrium.
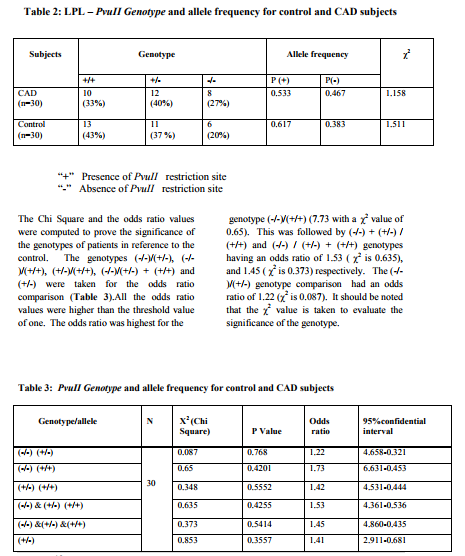
 The statistical reports for CAD and control subjects were shown in table 4 and 5. In control and CAD subjects, there were no significant differences in total cholesterol, HDL and TG level in common genotypes in Pvu II RFLPs. The total cholesterol, TG and HDL levels were found to be highest in the +/- genotype in patients and +/+ genotype in control. The LDL level was found to be higher in the +/- genotype, both in patients and control.
The statistical reports for CAD and control subjects were shown in table 4 and 5. In control and CAD subjects, there were no significant differences in total cholesterol, HDL and TG level in common genotypes in Pvu II RFLPs. The total cholesterol, TG and HDL levels were found to be highest in the +/- genotype in patients and +/+ genotype in control. The LDL level was found to be higher in the +/- genotype, both in patients and control.

DISCUSSION
Lipoprotein lipase is a candidate gene for CAD risk, and the possible association of the LPL – Pvu II polymorphism to CAD was studied. The genotype distribution of LPL polymorphism at a Pvu II polymorphic site was investigated. The results of various association studies of LPL – Pvu II polymorphisms with CAD have been inconsistent. Some previous studies have defined, an association between the extent of CAD and the LPL – Pvu II (+/+) genotype and the LPL – Pvu II (-/-) genotype to be moderately associated with CAD. Other studies did not define any significant difference in the distribution of LPL – Pvu II polymorphism between the healthy group and the CAD groups suggesting lack of association between any of the LPL Pvu II genotypes and CAD. The relevance of the Pvu II genotypes may vary among different populations. In the present study, the distribution of the LPL – Pvu II genotypes was significantly different in CAD and control study groups; the frequency of the +/- and -/- genotypes were higher in patients than in controls. Analysis of intra genotype variance of mean values of lipid levels showed that variability of Pvu II in LPL contributes to a certain extent to the level and variability in serum total cholesterol and LDL cholesterol levels. The LPL Pvu II genotypes, the CAD subjects heterozygous (+/-) for the higher total cholesterol, triglycerides, HDL and LDL cholesterol compared with homozygous +/+ and -/- subjects. The findings indicates no significant differences between the prevalence rates for the LPL – Pvu II genotypes in both the study groups, suggesting a very modest association between any of the LPL – Pvu II genotypes and CAD . This study was cross – sectional raising the possibility of changes in prevalence of LPL polymorphisms among cases and control subjects due to differential survival rates for CAD patients presenting for angiography based on LPL genotype status. This seems unlikely, but can be addressed by prospective studies. Disease and control groups differed in some baseline variables, as expected. However, when differences were accounted for by conditional multivariate logistic regression, the relative risk associated with the polymorphism was maintained or augmented.
References:
1. Chamberlain,JC.,Thorn,JA.,Oka,,K.,et al.(1989).DNA polymorphism at the LPL gene:associations in normal and hypertriglyceridemic subject. Atherosclerosis 79, 85-9.
2. Cryer,A.(1981).Role of LPL in lipdd metabolism.International Journal of Biochemistry 13,525-41.
3. Deep,SS.,Peng,R.(1989).Structure of the LPL gene.Biochemistry 28,4131-35. 4. Eckel,RH.,(1989).LPL a multifunctional enzyme relevant to common metabolic disease.The New England Journal of Medicine 320,1060-32.
5. Fisher,KL.,Fitzgerald,GA.,Lawn,RM.(1 987).Two polymorphisms in the human LPL gene.Nucleic Acids Research 15,7657.
6. Godota,T.,Yamada,N.,Murase,T.,Shima noh.,et al.(1992).Detection of three separate DNA polymorphisms in the human LPL gene by amplification and restriction endonuclease digestion.Journal of Lipid Research 33,1067-72.
7. Heinzmann,C.,Ladias,J.,Antonarakis,S., Kirchgessner,T.,et al.(1987).RFLP for the human LPL gene-HindIII.Nucleic Acids Research 15,6763.
8. Heizmann,C.,Kirchgessner,T.,Kwiterovi ch,PO.,Ladias,JA.,et al.(1991)..DNA polymorphism haplotypes of the human LPL gene;possible association with high density lipoprotein levels.Human Genetics 86,578-84.
9. Jemaa,R.,Tuzet,S.,Portos,C.,Betoulle,D., et al.(1995).LPL gene polymorphisms in association with hypertriglyceridemia and body mass index in obese people.Journal of Obesity Related to Metabolic Disorders 19,270-74.
10. Lusis,AJ.(2000).Atherosclerosis.Nature 407,233-41.
11. Murthy,V.,Julian,P.,GagneC.(1996).Mol ecular pathology of the human LPL gene.Pharmacol Therapy 70,101-35.
12. Oka,K., Tkalcevic,GT, Stocks,J., Galton,Dj.(1989). Nucleotide sequence of PvuII polymorphic site at the human LPL gene.Nucleic Acids Research 17, 6752.
13. Olivercrona,T., Hultin,M., Bergo,M. and Olivercrona,G. (1997). Lipoprotein lipase:regulation and role in lipoprotein metabolism.Proc Nutural Society 56,723-9.
14. Wang, XL., Mc Credie, RM., Wilken, DE.(1996). Common DNA polymorphism at the LPL gene association with severity of CAD and diabetes.Circulation 93,1339-45.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





