IJCRR - 4(18), September, 2012
Pages: 24-30
Date of Publication: 29-Sep-2012
Print Article
Download XML Download PDF
DEVELOPMENT OF ASSAY METHOD FOR ARTESUNATE BY INDIRECT SPECTROPHOTOMETRY OF ITS ALKALINE DEGRADATION PRODUCT(S)
Author: Nwodo, NJ Nnadi, CO, Okoye, FBC
Category: Healthcare
Abstract:Artesunate contains lactone ring known to degrade in alkaline medium; a reaction that could serve as a novel, simple, precise and rapid spectroscopic tool for quantitative determination in bulk and dosage forms. The degradation of artesunate in nucleophilic agents was investigated. Absorption spectra of artesunate in ethanol, Britton-Robinson buffer, and ammonium buffer (pH 5-12) were obtained. The effects of time and temperature on the stability of the degradation products were also evaluated. Each change in pH caused the spectrum of artesunate to change significantly with time up to pH 12, where a prominent peak at 225 nm was observed. Artesunate follows linearity in the concentration range of 2-20 \?g/ml with correlation coefficient of 0.999 in the proposed method. At constant temperature, therefore, the degradation products of artesunate at pH 12 could be harnessed for qualitative and quantitative determination of artesunate in dosage forms and biological fluids. The method was validated for specificity, precision and accuracy.
Keywords: Absorption maxima, pH, Buffer solution, Validation
Full Text:
INTRODUCTION
Malaria poses a huge economic burden on countries where the disease is endemic. According to the World Health Organization (WHO), malaria is a significant public health problem in more than 90 countries inhabited by some 2400 million people and causes an estimated 300 million acute illness each year with at least a million deaths1 . More than 90 % of these occur in Sub-Saharan Africa, and it is estimated that the disease kills an African child every 30 seconds2 . Spread and emergence of old and new drug-resistant parasites is threatening malaria control programmes in most malaria-endemic regions3 . The high morbidity of malaria necessitated the use of many antimalaria drugs in therapy, at least to control or eradicate the disease4 . One of such drugs is artesunate, a qinghaosu-based compound derived from the herb artemisia. It is a fine white crystalline powder, slightly soluble in water and freely soluble in ethanol.
Due to the extensive use of artesunate alone or in combination as artemisinin-based combination therapy ( ACT) in malaria treatment, various assay methods have been developed5,6,7. Unfortunately, these methods are costly, time consuming and non-environmental friendly. The International Conference on Harmonisation (ICH) guidelines for development of assay methods for drugs require the consideration of the regulatory status of the developed methods of assay, review of literature on stability-indicating assay8 , assessment of the extent to which the developed method meet current regulatory requirement, techniques employed in development of assay methods and development of validated stability-indicating assay methods (SIAMs) that are likely to meet regulatory requirement9 . Studies of this nature provides a finger-print for spectroscopic detection of artesunate indirectly by understanding the reaction of the quinghaosu backbone of artesunate. Quinghaosu is unstable in alkaline medium and may decompose at room temperature to form many degradation products such as octahydro-indene10. The cleavages of the lactone ring and/or epoxide linkage and succinate moiety of artesunate can be harnessed spectrophotometrically to identify the drug qualitatively and quantitatively in non-interfering matrices such as dosage forms and biological fluids.
MATERIALS AND METHODS
The major materials used for the study are Artesunate (Swiss Pharma., Nigeria), Ultraviolet/Visible Spectrophotometer (Model UNICO 2100, China) and pH meter (Eutech, Japan). All reagents for this study were used as received without further purification.
(a) Preparation of Buffer Solutions
0.4 M each of orthophosphoric acid, boric acid, acetic acid, sodium hydroxide, ammonium hydroxide, ammonium chloride and 2.0 M ammonium hydroxide were prepared. BrittonRobinson (BR) buffer (pH 5-8) and ammonium buffer (pH 8-12) were prepared as described11 .
(b) Preparation of Standard Solution of Artesunate in Ethanol
A 100 µg/ml artesunate solution was prepared by accurately weighing and dissolving 100 mg of pure artesunate powder in ethanol. The solution was made up to 1 litre with ethanol. From the stock solution, 10 µg/ml solution was prepared by serial dilution with ethanol and scanned in UV spectrophotometer against ethanol blank.
(c) Method Development
(i) Degradation Analysis of Artesunate
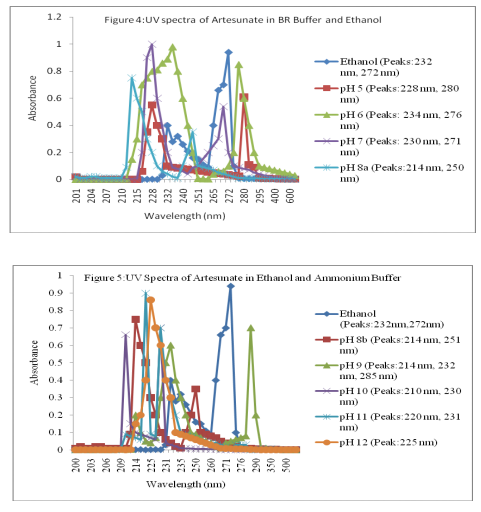
A 10 µg/ml solution of artesunate was prepared from the standard solution using the BR buffer at pH 5 by serial dilution. The absorption spectrum was obtained against ethanol-BR solution blank. The procedure was repeated using BR buffer at pH 6-8 and ammonium buffer 8-12. The spectra of the drug were shown in Figures 4 and 5.
(ii) Effect of Time and Temperature on the Stability of Degradants.
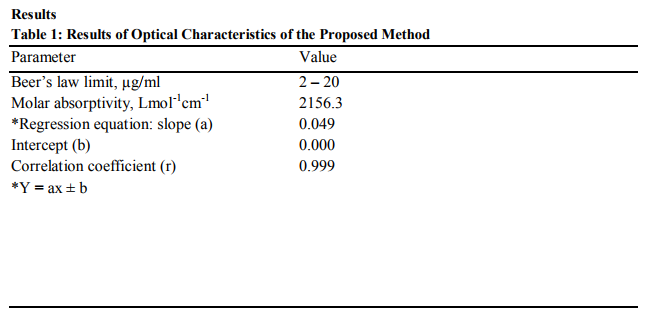
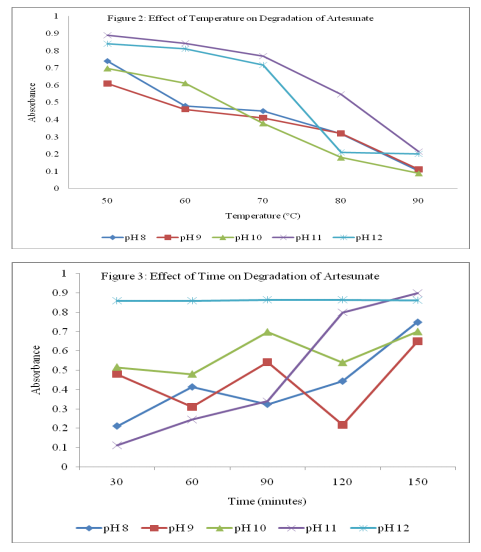
A 10 µg/ml solution of artesunate was prepared from the stock solution using ammonium buffer (pH 8) and the absorbances were taken after 30, 60, 90, 120 and 150 minutes of standing. The absorbances of fresh solution were taken after heating to 50, 60, 70, 80 and 90 ºC at ?max specific for each pH. These procedures were repeated for ammonium buffer (pH 9-12). Plots of the absorbance against time and temperature were shown in Figures 2 and 3 respectively.
(iii) Calibration Curve of Artesunate at Degradation Conditions.
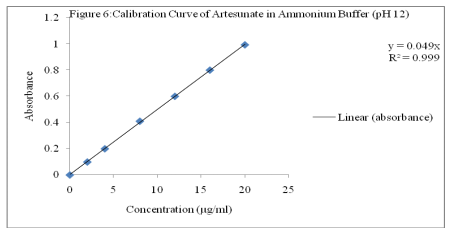
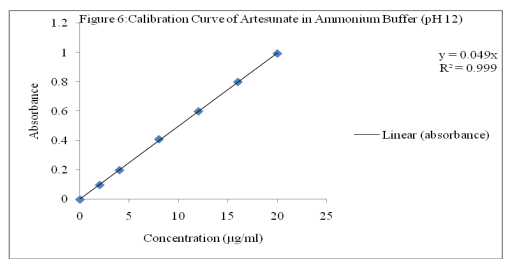
Aliqouts of stock solution were further diluted with ammonium buffer (pH 12) to get working solution of 2, 4, 8, 12, 16 and 20 µg/ml. Subsequently, the absorbances were taken at ?max of 225 nm against ethanol-buffer blank. A calibration curve of the absorbance against concentration was plotted as shown in Figure 6.
(iv) Assay Procedure for Formulations
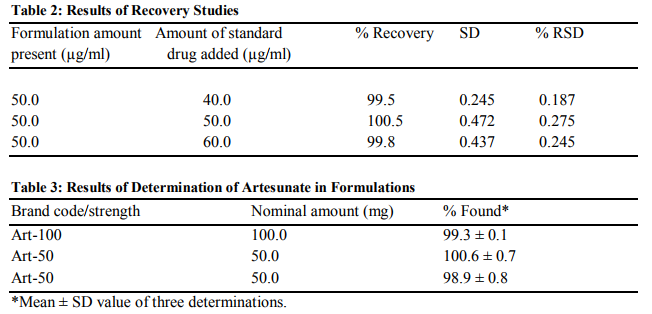
An amount of finely ground tablet powder equivalent to 50 mg of artesunate was accurately weighed into a 100 ml calibrated flask, 50 ml of ethanol added and shaken for 10 minutes. Then, the volume was made up to the mark with ammonium buffer (pH 12), mixed well, and filtered using a Whatman No. 42 filter paper. First 10 ml portion of the filtrate was discarded and a suitable aliquot of the subsequent portion 500 μg/ml was diluted appropriately to get concentrations for analysis by the method. The results are shown in Table 3.
(d) Method Validation
(i) Precision
The precision of the developed method was determined by measuring the absorbances of six separate samples containing known amount of drug prepared under degradation condition. The method was validated by calculating the slope, intercept, correlation coefficient and optical characteristics (ICH guidelines 1995) as shown in Table 1.
(ii) Recovery Studies
Recovery studies were carried by adding 80, 100 and 120 % of pure artesunate to different samples of tablet powder containing equivalent of 50 mg of artesunate12. From the amount of drug found, percentage recovery was calculated. The percentage recovery and the % RSD values are shown in Table 2.
DISCUSSION
The proposed method is based on the degradation of the lactone ring of the artesunate in alkaline medium and the reaction is followed spectrophotometrically for quantization purposes13. The UV absorption spectra of artesunate in ethanol and buffer solution are illustrated in Figures 4 and 5 for buffers of pH 5-8 and pH 8-12 respectively. The peak at 232 nm obtained in ethanol could be attributed to succinate moiety of artesunate while the peak at 272 nm could be due to lactone ring11. The minor changes in the maximum wavelength of absorption observed for pH 5-7 suggest the introduction of functional groups in the course of the degradation. The bathochromic shift of peak at 272 nm to 280 at pH 5 may be attributed to the cleavage of endoperoxide linkage of artesunate leading to addition of C=C bond5 . The appearance of exactly same spectra for pH 8a and 8b shows that buffer system has no interference in the degradation of artesunate. It was observed that different degradation products were formed in all the buffer solution used but only the products formed at pH 12 was stable on standing. Assuming that absorbance is proportional to concentration, as stated in BeerLambert’s law, the the decrease in absorbance as temperature increases indicates a decrease in the concentration of the product. This signifies that the product is not very stable to temperature. At constant temperature, therefore, the product can be said to be stable as the variation in the absorbance was not pronounced as shown in Figures 2 and 3. The reliability, accuracy and validity of the proposed method were ascertained by carrying out recovery studies. The recovery of pure drug added was quantitative and revealed that co-formulated substances (excipients) did not interfere in the determination as shown in Table 2. This further stresses the selectivity of the method for analysis of artesunate. In order to check the validity of the proposed method, analysis of artesunate in some commercial formulations was carried out. Table 3 shows the results of studies in which there is close agreement between the result obtained by the proposed method and label claims indicating high accuracy and precision. The proposed method is simple, precise and useful in routine determination of artesunate in pharmaceutical dosage forms and biological fluids. Research is currently in progress to isolate, characterize and elucidate the structures of the degradation product(s) of artesunate for more accurate on-the-spot detection of the drug.
CONCLUSION
A method for indirect determination of artesunate in bulk and dosage forms has been developed and validated in the concentration range of 2-20 µg/ml (R2 = 0.999) with average percentage recovery of 99.93 from tablets. The developed method is selective, novel, precise, linear, simple accurate and suitable for routine determination of artesunate without interference from dosage matrices.
ACKNOWLEDGEMENT
The authors are grateful to Chidera Charles-Nnadi for her technical support and to the Stephen Oluwole Awokoya Foundation for Science Education, Lagos Nigeria for providing financial assistance. Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to authors/editors/publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed.
References:
1. World Health Organization Global Report on Antimalaria Drug Efficacy and Drug Resistance. WHO Geneva, 2010.
2. Range HP, Dale MM, Ritter JM, Flower RJ. Range and Dale’s Pharmacology. Elsevier 6(1) China. 2007; 467-474.
3. Kamau E, Alemayehu S, Feghali KC, Tolbert LS, Ogutu B, Ockenhouse CF. Development of a TaqMan Allelic Discrimination Assay for Detection of single nucleotides polymorphisms associated with anti-malarial drug resistance. Malaria Journal. 2012; 11:23 doo, 10.1186/1475-2875-11-23. http//www.malariajournal.com/content/11/1/2 3.
4. Boulangier D, Dieng Y, Cisse B. Antischistosomal Efficacy of artesunate combination therapies administered as curative treatment for malaria attacks. Tropical Med. Journal. 2007; 10(102): 198- 265.
5. Thekke VS, Badiadka N. A Simple and Rapid Spectrophotometric Method for the Determination of Artesunate in Pharmaceuticals. Eurasian J. Anal. Chem. 2009; 4(1): 119-126.
6. Esimone CO, Omeje EO, Okoye FBC, Obonga WO, Onah BU. Evidence for the spectroscopic determination of Artesunate in dosage form. J Vector Borne Dis 45, pp. 2008; 281–286.
7. Olajire AA, Olakunle S.I, Oluwafunmibi PD, Olutayo SO. Derivatization of artemisinin derivatives using 4-carboxyl -2, 6-dinitro benzenediazonium (CDNBD) ion Acta Pharmaceutica Sciencia. 2010; 52: 269-280.
8. . Monika B, Saranjit S. Development of validated stability-indicating assay methods: critical review. Journal of Pharmaceutical and Biomedical Analysis 2002; 28:1011–1040.
9. Stability Testing Of New Drug Substances And Products Q2 (R1) Nov. 2005; ICH Harmonized Tripartite Guideline.
10. Liu JM, Ni MY, Fan YF, Tu YY, Wu ZH, Wu YL, Chou WS. Structure and Reactions of Artemisinin. Huaxue Xuebao, 1979; 37(2): 129–141.
11. Viviane MI, Leticia NCR, Claudia CIG, Maria VBZ. Spectrophotometric determination of Cefaclor in Pharmaceutical Preparation. Quimica Nova 1998; 22(2): 801-807.
12. Jeswani RM, Sinha PK, Topagi KS, Damle MC. A Validated Stability Indicating HPTLC Method for Determination of Cephalexin in Bulk and Pharmaceutical Formulation International Journal of PharmTech Research. 2009; 1(3): 527-536.
13. Basavaiah K, Anil Kumar UR. Simple Spectrophotometric Methods for the Determination of Zidovudine in Pharmaceuticals Using Chloramine-T, Methylene Blue and Rhodamine-B as Reagents. E-Journal of Chemistry. 2006; 3(12):173-181.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





