IJCRR - 4(2), January, 2012
Pages: 21-31
Print Article
Download XML Download PDF
CLICK CHEMISTRY: AN APPROACH TO SUSTAINABLE DEVELOPMENT IN DRUG SYNTHESIS
Author: Abhishek Kumar, Pankaj Kumar, Himanshu Joshi
Category: General Sciences
Abstract:Click chemistry is a chemical philosophy introduced by K. Barry Sharpless in 2001 and describes chemistry tailored to generate substances quickly and reliably by joining small units together. This is inspired by the fact that nature also generates substances by joining small modular units. Click chemistry is not a specific reaction; it is a concept that mimicks nature. Sustainable development requires redesigning many organic chemical processes, most of which are often based on technology developed in the first half of the 20th century, and inventing new reactions that use and produce safer chemicals under more environmentally benign conditions. Click Chemistry is often cited as a style of chemical synthesis that is consistent with the goals of green chemistry. The focus is on minimizing the hazard and maximizing the efficiency of any chemical choice. Moreover Sharpless notes that ?many of the reactions that meet the click
chemistry standard often proceed better (faster and more selectivity) in water than in organic
solvents. Click Chemistry requires simple reaction conditions, preferably solvent free or water
as the solvent which results in generation of inoffensive by-products. Indeed it has been found
that most of the reactions that meet click chemistry standards often proceed rapidly in water
and with high yield than in an organic solvent. The aim of the present article is to provide in
detail the click chemistry reactions using solvent free or water as the solvent that is consistent
with the goals of sustainable chemistry.
Keywords: Click chemistry, green chemistry, sustainable, solvent free, water
Full Text:
INTRODUCTION
Sustainable development requires redesigning many organic chemical processes, most of which are often based on technology developed in the first half of the 20th century, and inventing new reactions that use and produce safer chemicals under more environmentally benign conditions1 . Water is generally considered as an ideal solvent in terms of the environmental impact and low cost. In addition, water is superb heat sink and thus can control the exothermic transformations without side reactions. The free energies of organic molecules are substantially greater when poorly solvated in water, and thus often result in increased reactivity which can compensate for low concentration of reactant molecules in water. Water can activate the electrophile and nucleophile through the formation of hydrogen bonding2 and also can differentiate the reactivities of competing nonpolarizable and polarizable species. The protection- deprotection of the functional groups such as –OH, -NH2, -COOH may not be necessary. Moreover the reaction products can sometimes be isolated simply by decantation or filtration. Due to these inherent features of water, it is natural outcome that water can be suitable solvent of choice for such designed set of click reactions. To date pericyclic, condensation, oxidation and reduction reactions are routinely carried out in aqueous medium3 . Indeed, it has been found that most of the reactions that meet click chemistry standards often proceed rapidly in water and with high yields than in an organic solvent. The simple protic functional groups like hydroxyl and amides which are ubiquitously present in biologically active organic molecules can be tolerated by using water as a solvent, which offers another significant advantage of performing click chemistry in water in perspective of drug discovery. The 1, 2-epoxide functionality is largely present in nature, is biologically important and is a powerful building block in organic synthesis4 . Recently Sharpless5 , following the chemical lead of mother nature, proposed to term ?click chemistry? the synthetic approach that generates substances ?by joining small units together with heteroatom links (C-X-C)? and defined the criteria that a process must meet to be useful in this context. A ?click reaction? that uses this strategy is the nucleophilic ring-opening of 1, 2- epoxides.Moreover Sharpless notes that ?many of the reactions that meet the click chemistry standard often proceed better (faster and more selectivity) in water than in organic solvents.. In this respect water is unique as reaction medium.? An overview of the click reactions carried in solvent free or water as the solvent has been presented. Water as a solvent for click chemistry The concept of green or sustainable chemistry was born around 1990 thanks to a small group of chemists who, ahead of the times, clearly saw that the need for more environmentally acceptable processes in chemical industry had to become a top priority in RandD activities. Over the past two years, we have found that many of the reactions that meet click chemistry standards often proceed better in water than in an organic solvent. This is a natural outcome of one or more of the following five factors. 1) Click reactions often proceed readily in hot water, to give a single product, even when one or more of the reactants, as well as the product, appear to be insoluble in this medium. The fact that reactions between organic species in aqueous solution can have higher apparent rate constants than the same processes in organic media has been observed and exploited by a number of laboratories6 . Among the many explanations offered for such phenomena, we call particular attention to the notion that the free energies of organic molecules are substantially greater when poorly solvated in water, and often impart increased reactivity, which compensates for the low concentration of the participants7 . 2) Nucleophile additions to epoxide8 (?homocarbonyl?) and aziridine (?homoimine?) electrophiles (as well as aziridinium and episulfonium ions) are favored by solvents best able to respond continuously to the demanding range of hydrogen-bonding situations that arise during these processes. In this respect, water is unique, and for the same reasons, it is the perfect milieu for reversible carbonyl chemistry. 3) Two important subsets of olefin and acetylene click reactions are oxidations by electrophilic reagents and cycloaddition reactions. These processes are either concerted or involve polarizable nucleophiles/electrophiles, so that water is not an interfering medium9 . More generally, it should be appreciated that the use of water offers the greatest leverage for differentiating the reactivities of competing ?hard? (nonpolarizable) and ?soft? (polarizable) species. 4) A highly favorable reaction of two solutes (say at 0.1M concentration) is usually much faster than a low driving force side-reaction of one of the solutes with solvent water (55 M). The Schotten - Baumann method for making amides from acyl or sulfonyl halides in water is a well known example given below.
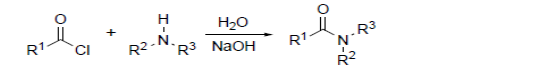
5) Water is a superb heat sink, due to its high heat capacity, and has a convenient boiling temperature; both are useful for large-scale processes. Water is usually regarded as an ideal solvent in terms of its environmental impact and low cost. A benefit which is little appreciated but has enormous consequences is that most hydroxy and amide groups will not interfere with click reactions performed in water. As a consequence, the installation and removal of protecting groups is avoided, probably the best single reason for adopting this style of synthesis. Indeed, we view many of the best reactions for installing protecting groups as good click reactions in their own right. These considerations highlight the fact that, although click reaction components are necessarily highly reactive, their chemoselectivity profiles are quite narrowly defined, that is, orthogonal to an unusually broad range of reagents, solvents, and other functional groups. This attribute allows for reliable and clean sequential transformations of broad scope10. For example, opening epoxide or aziridine rings by HN3 installs a highly reactive ?sticky spot? for [3+2] cycloaddition with alkynes, but one that is invisible to most other functional groups. The types of reactions, and especially reaction conditions, which fall into the click chemistry category were more common in the organic literature of 50 to 100 years ago. Few solvents were used then, and heat was the preferred way to speed up reactions. The dearth of available purification techniques meant that processes were chosen for their reliability in giving a single isolable product.
Catalysis
Adopting a K. B. Sharpless statement11 , catalysis is the engine that drives the development of chemistry. Everybody can easily recognize that top achievements in applied chemistry are focused on industrial applications of catalysis, rational design, serendipitous discovery or combinatorial identification of new ligands, catalysts, new solid supports (organic, inorganic, amorphous or mesoporous silica phases, metal organic frameworks, etc). An ideal catalyst should approach 100% selectivity while reaching high levels of productivity. Selectivity refers first of all to (i) chemoselectivity, which means the catalyst must be able to select preferred reactants from complex mixtures, (ii) regioselectivity, which means selection of preferred sites of the reacting substrate and (iii) stereoselectivity, which means preferred formation of a single stereoisomer.
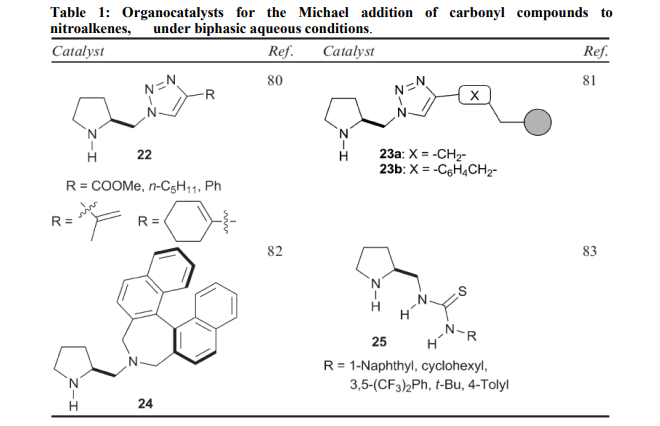
Based on ??click chemistry‘‘, pyrrolidine catalysts such as 22, containing a triazole ring, have been shown to efficiently promote a highly diastereoselective and enantioselective Michael addition of ketones to nitroalkenes12. The triazole moiety is essential to promote the reaction in water with excellent yields and enantiocontrol. A solid-phase version, 23, has also been proposed. Here the1, 2, 3- triazole ring, constructed through a click 1, 3-cycloaddition, also grafts the chiral pyrrolidine monomer onto the polystyrene backbone and provides a structural element capable of conferring the catalyst with high catalytic activity and enantioselectivity, particularly in the case of ketones as donors. Catalytic performance is optimized by using water and DiMePEG as an additive. A basic advantage is the easy recyclability13 . With chiral diamine 24, in the form of a trifluoroacetate salt, the classic aqueous biphasic protocol has been successfully applied to the asymmetric Michael reaction of ketones with both aryl and alkyl nitro olefins. Brine is used as the aqueous phase14 . The thiourea functionality, inserted on the most frequently used chiral pyrrolidine scaffold, works excellently as reactivity and enantioselectivity control co-factor by chelating the nitro group of the acceptor. This solution, adopted in 25, provides a family of robust catalysts that afford high yields (up to 98%) and great stereoselectivities (up to 99: 1 dr and 99% ee) in direct Michael additions of ketones to various nitroolefins in water15 . Reactions in the presence of metal salts Azidolysis of 1, 2-epoxides is a widely investigated organic reaction because 1, 2- azidoalcohols are precursors of vicinal amino alcohols and are building blocks for carbohydrates and nucleosides16 . The classical protocol uses NaN3 (5 mol/eq) as reagent in the presence of NH4Cl (2.3 mol/eq) as coordinating salt in alcohol-water at 70-80 °C. Some examples are illustrated in Table 217 and Scheme 118, 19
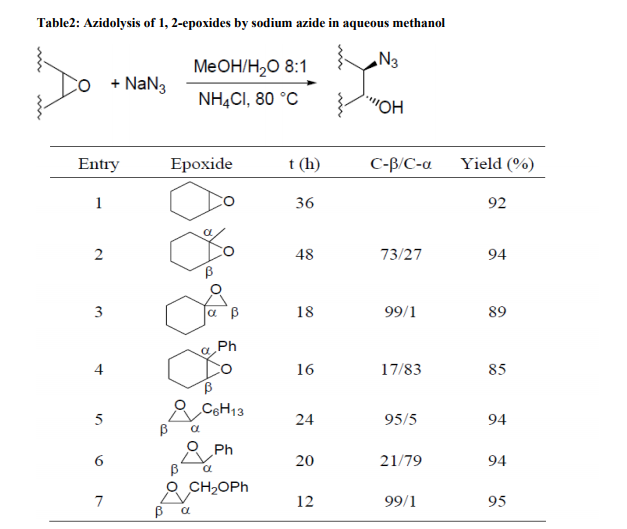
Under these conditions the reaction is completely anti-stereoselective and generally requires a long reaction time. The attack of nucleophile on substituted oxirane ring occurs mainly on the least substituted carbon except when the substituent is an aryl group.
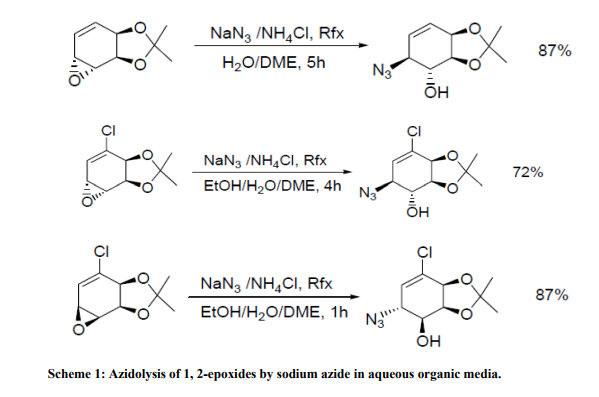
In this case the nucleophilic attack generally occurs mainly on the benzylic carbon (Table 2, entries 4 and 6). In the absence of a specific substituent on the α-and β-carbons of the oxirane ring, the nucleophile preferentially attacks the carbon which is less influenced by unfavorable effects of electron-withdrawing functionalities present in the molecule (Scheme1).
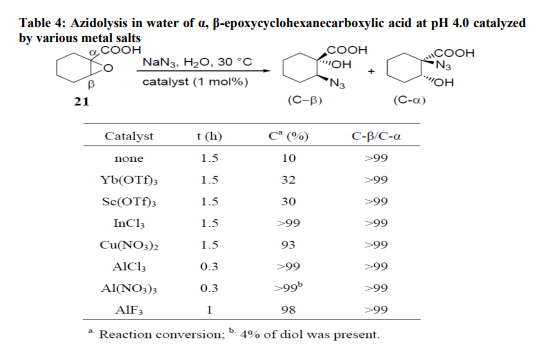
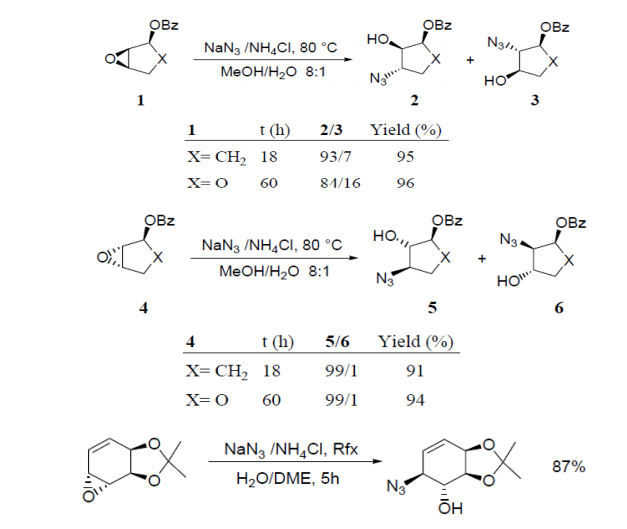
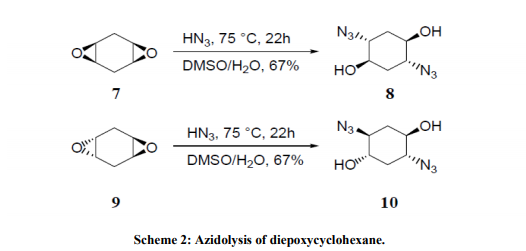
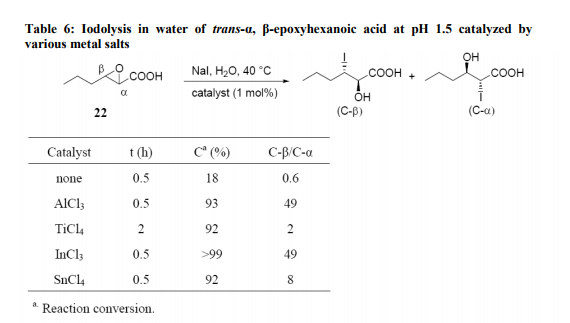
Azidolysis of cis- and trans- 1, 4- diepoxycyclohexanes 7 and 9 (Scheme 2) with hydrazoic acid, generated in situ from NaN3 and p-toluenesulfonic acid, carried out in 1:1 DMSO/H2O mixture at 70°C, gave the azidoalcohols 8 and 10, respectively, as sole products which were converted to corresponding aminocyclitols with good yields by catalytic reduction20 . The reason for using an aqueous-organic medium for the azidolysis of 1, 2-epoxides is to carry out the reaction under homogeneous conditions solubilizing both the sodium azide (water) and the epoxide (organic solvent). Recently we have shown21 that the azidolysis of 1, 2-epoxides can be suitably performed in water alone under heterogeneous conditions. The nucleophilic addition was totally antidiastereoselective and the reactivity and regioselectivity of the process and the competition of the azido ion with the water or with the hydroxide ion were controlled by working at suitable pH values. Some results are reported in Table 3. The conversion of 1, 2- epoxide into azidoalcohol was quantitative. With highly hydrophobic epoxides, the azidolysis was accelerated by carrying out the reaction in the presence of cetyltrimethylammonium bromide (CTABr). Metal Salt Catalyzed Reactions Metal salt-promoted nucleophilic ringopening of 1, 2-epoxides were investigated in an attempt to improve the regioselectivity and the reaction rate attained by using classical conditions (NaN3/NH4Cl, in alcohol or alcohol/water at 70-80 °C). Most of the work was carried out inorganic solvent using a variety of metal salts (LiClO4, LiOTf, Mg(ClO4)2, Zn(OTf)2, NaClO4, KClO4, Ti(O-i-Pr)4),22,23 generally in large excess. Lewis acidcatalyzed reactions carried out in just water have been recently investigated focusing on the azidolysis24, iodolysis25, bromolysis and thiolysis26 of 2,3-epoxycarboxylic acids and their esters to develop simple one-pot procedures in aqueous media for the synthesis of hydroxyamino acids, hydroxyacids, and hydroxysulfides.
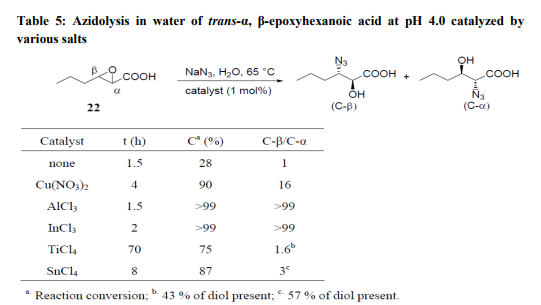
The azidolysis reaction of α, β- epoxycyclohexane carboxylic acid (21) (Table 4) was efficaciously catalyzed in water at pH 4.0 and 30°C by 1 mol% of Cu(NO3)2, InCl3, AlCl3, Al(NO3)3 and AlF3. These salts, with the exception of Cu(NO3)2, were not operative at pH 7.0. Yb(OTf)3, and Sc(OTf)3 were not efficient at either pH 4.0 or at pH 7.0. The reactions were always highly C-β-regioselective. The catalytic effect of various Lewis acids on the azidolysis of trans- α, β- epoxyhexanoic Acid (22) was also investigated. Cu(NO3)2, AlCl3 and InCl3 were again the best catalysts at pH 4.0, while SnCl4 showed little activity and TiCl4 slowed the reaction rate; both these salts gave mixtures of C-β and C-α adducts along with large amounts of diols (Table 4).
CONCLUSION
The present article describes the click chemistry reactions performed in solvent free or water as the solvent which is consistent with the goals of green chemistry. It emphasizes the advantage of using water as a solvent in click chemistry. It describes the nucleophilic ring-opening of 1, 2-epoxides in aqueous media and the role of organocatalysts in click chemistry reactions. Many of the reactions that meet the click chemistry standard often proceed better (faster and more selectivity) in water than in organic solvents.
ACKNOWLEDGEMENT
Authors acknowledge the immense help received from the scholars whose articles are cited and included in references of this manuscript. The authors are also grateful to authors/editors/publishers of all those articles, journals and books from where the literature for this article has been reviewed and discussed.
References:
1. Tundo P, Anastas PT. Green Chemistry: Challenging Perspectives. Oxford University Press 2000.
2. Chakraborti et al. Green Chem 2007; 9:11(advance article).
3. Li CJ, Chang TH. Organic reactions in aqueous media. Wiley NY 1997.
4. Fringuelli F, Piermatti O, Pizzo F. Trends in Organic Chemistry. Res Trends Pub 1997; 6:181.
5. Kolbe HC, Finn MG, Sharpless KB. Angew Chem Int Ed 2001; 40: 2005.
6. Breslow R. Pure App Chem 1998; 64:6094-96.
7. Gajewski JJ. Acc Chem Res 1997; 30:219-225.
8. Fringuelli F, Piermatti O, Pizzo F, Vaccaro L. J Org Chem 1999; 64:6094- 96.
9. Mersbergen DV, Wijnen JW, Engberts JBFN. J Org Chem 1998; 63:8801- 8805.
10. Tietze LF, Modi A. Med Res Rev 2000; 20:304-322.
11. Colacino E, Nun P, Maria FM, Martinez J, La
12. Kumamoto K, Ichikawa Y, Kotsuki H. Synlett 2005; 14:2254.
13. Matsumoto K, Hashimoto S, Uchida T, Iida H, Otani S. Chem Exp 1993; 8:475.
14. Sugihara T, Takebayashi M, Kaneka C. Tetrahedron Lett 1995; 36:5547.
15. WG Dauben, Hendrcks RT. Tetrahedron Lett 1992; 33:603.
16. Jacobs GA, Tino JA, Zahler R. Tetrahedron Lett 1989; 30:6955.
17. Chini M, Crotti P, Macchia F. Tetrahedron Lett 1990; 31:5641.
18. Crotti P, Di Bussolo V, Favero L, Macchia F, Pineschi M. Eur J Org Chem 1998; 1675.
19. Banwell MG, Haddad N, Hudlicky T, Nugent TC, Mackay MF, Richards SL. J Chem Soc 1997; 1:1779.
20. Kavadias G, Droghini R. Can J Chem 1979; 57:1870.
21. Fringuelli F, Germani R, Pizzo F, Santinelli F, Savelli G. J Org Chem 1992; 57:1198.
22. Chang JM, Sharpless KB. J Org Chem 1985; 50:1560.
23. Azzena F, Crotti P. Favero L, Pineschi M. Tetrahedron 1995; 51:13409.
24. Fringuelli F, Pizzo F, Vaccaro L. Synlett 2000; 311.
25. Amantini D, Fringuelli F, Pizzo F, Vaccaro L. J Org Chem 2001; 66:4463.
26. Fringuelli F, Pizzo F, Tortoioli S, Vaccaro L. Adv Synth Catal 2002; 344-379.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





