IJCRR - 4(8), April, 2012
Pages: 124-146
Date of Publication: 25-Apr-2012
Print Article
Download XML Download PDF
BIOEQUIVALENCE AND HIGHLY VARIABLE DRUGS: AN OVERVIEW
Author: Vikram Lohar, Harsh Patel, Arvind Singh Rathore, Sandeep Singhal, Ashish Kumar Sharma, Parul Sharma
Category: Healthcare
Abstract:Bioequivalence studies are the preliminary requirement for generic products to enter in the market. The manufacturer (generic) must be in limit with that of innovator (branded) formulation (reference listed drug) within the limits approved by respective governing bodies. As per biopharmaceutical classification system the drugs falls in the category I to IV on the basis of permeability and solubility data. Drugs belonging to the category of poor solubility and poor permeability data uphold bioequivalence issues. Due to this high variability, large sample size may be needed in BE studies to give adequate statistical power to meet FDA BE limits, and thus designing BE studies for HVDs is challenging. Consequently
development of generic products for HVDs is a major concern for the generic drugs industry. Major regulatory agencies also considered different approaches for evaluating BE of highly variable drugs. From 2004 onward the FDA started looking for alternative approaches to resolve this issue, and eventually found that replicate crossover design and scaled average BE provides a good approach for evaluating the BE of highly variable drugs and drug products as it would effectively decrease sample size, without increasing patient risk.
Keywords: Bioequivalence, Highly Variable Drugs, Pharmacokinetic.
Full Text:
INTRODUCTION
Generic drug According to the U.S. Food and Drug Administration (FDA), generic drugs are identical or within an acceptable bioequivalent range to the brand name counterpart with respect to pharmacokinetic and pharmacodynamic properties. By extension, therefore, generics are considered (by the FDA) identical in dose, strength, route of administration, safety, efficacy, and intended use. The FDA‘s use of the word identical is very much a legal interpretation, and is not literal. In most cases, generic products are available once the patent protections afforded to the original developer have expired. When generic products become available, the market competition often leads to substantially lower prices for both the original brand name product and the generic forms.
Hatch Waxman Act
Using bioequivalence as the basis for approving generic copies of drug products was established by the "Drug Price Competition and Patent Term Restoration Act of 1984,? also known as the Waxman-Hatch Act. Under Hatch-Waxman Act, one of the following four certifications has to be made while filing an ANDA: [Food and drug administration, center for drug evaluation and research (CDER)]. 1 Bioavailability (BA) and bioequivalence (BE) studies provide important information in the overall set of data that ensure the availability of safe and effective medicines to patients and practitioners. BA and BE measures are frequently expressed in systemic exposure measures, such as area under the plasma concentration-time curve (AUC) and maximum concentration (Cmax). These measures of systemic exposure are assumed to relate in some way to safety and efficacy outcomes that may be expressed in biomarkers, surrogate endpoints, or clinical benefit end points. 2
Bioequivalence (BE) is defined as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study 3 . BE studies of systemically absorbed drug products are generally conducted by determining pharmacokinetic endpoints to compare the in vivo rate and extent of drug absorption of a test and a reference drug product in healthy subjects. A test product is considered bioequivalent to a reference product if the 90% confidence intervals for the geometric mean test/reference ratios of the area under the drug‘s plasma concentration versus time curve (AUC) and peak plasma concentration (Cmax) both fall within the predefined BE limits of 80–125% .4 The width of the 90% confidence interval is proportional to the estimated drug variability (in particular, within-subject variability for a crossover design) and inversely proportional to the number of subjects participating in the study. The BE limits of 80–125% are currently applied to almost all drug products regardless of the size of within-subject variability. As a result, the number of subjects required for a study of highly variable drugs or drug products can be much greater than normally needed for a typical BE study. For example, to demonstrate BE with 90% power, it was estimated that 136 subjects would be required for a drug with 60% within subject coefficient of variation even if the test and reference products were identical. 5
Traditional Bioequivalence Method
For systemically available drug products, FDA generally asks applicants to conduct BE studies with pharmacokinetic endpoints using a single dose, crossover design in healthy subjects. The processes of study design and workflow of BA/BE studies are presented in brief in Figure 1 and Table 1 describes various study designs generally used for BA/BE studies. Subjects receive a single dose of test and reference products on separate occasions with random assignment to the two possible sequences of product administration. Treatments are separated by a washout period of adequate duration such that the drug of interest can no longer be detected in plasma. The FDA generally asks applicants to conduct single dose studies rather than multiple dose studies because single dose studies are generally more sensitive to detecting potential differences between products 4 . For a product with multiple strengths, the highest strength is used in the BE study, unless precluded for reasons of safety. The number of subjects in the study should be sufficient to ensure adequate statistical power;most studies enroll from 24 to 36 subjects. The bioequivalence parameters AUC and Cmax are statistically analyzed using the two one-sided tests procedure to determine whether the average values for the measures estimated after administration of the test and reference products are comparable.6 This approach involves the calculation of a 90% confidence interval for the ratio of the averages of the measures for the test and reference products. 7 The choice of the current 80 to 125% acceptance limits for BE has been based on expert medical judgment and FDA experience with thousands of drug products that a difference of less than 20% in drug exposure was not clinically significant for most drugs.8 The 80% limit indicates that the test product is no less than 80% of the reference, while the 125% limit indicates that the reference product is no less than 80% of the test product (a 4:5 reference to test ratio is a 5:4 test to reference ratio).
ASSESSMENT OF BIOEQUIVALENCE
The assessment of BE of different drug products is based on the fundamental assumption that two products are equivalent when the rate and extent of absorption of the test/generic drug does not show a significant difference from the rate and extent of absorption of the reference/brand drug under similar experimental conditions as defined. As per the different regulatory authorities, BE studies are generally classified as:
1. Pharmacokinetic endpoint studies.
2. Pharmacodynamic endpoint studies.
3. Clinical endpoint studies.
4. In vitro endpoint studies.
The general descending order of preference of these studies includes pharmacokinetic, pharmacodynamic, clinical, and in vitro studies.
Pharmacokinetic endpoint studies
"These studies are most widely preferred to assess BE for drug products, where drug level can be determined in an easily accessible biological fluid (such as plasma, blood, urine) and drug level is correlated with the clinical effect. The statutory definition of BA and BE, expressed in rate and extent of absorption of the active moiety or ingredient to the site of action, emphasizes the use of pharmacokinetic measures to indicate release of the drug substance from the drug product with absorption into the systemic circulation. Regulatory guidance recommends that measures of systemic exposure be used to reflect clinically important differences between test and reference products in BA and BE studies. These measures include i) Total exposure (AUC0–t or AUC0–∞ for singledose studies and AUC0–τ for steady-state studies), ii) Peak exposure (Cmax), and iii) Early exposure (partial AUC to peak time of the reference product for an immediate-release drug product). Reliance on systemic exposure measures will reflect comparable rate and extent of absorption, which, in turn, will achieve the underlying goal of assuring comparable therapeutic effects. Single dose studies to document BE were preferred because they are generally more sensitive in assessing in vivo release of the drug substance from the drug product when compared to multiple dose studies. The following are the circumstances that demand multiple-dose study/steady state pharmacokinetics:
- Dose- or time-dependent pharmacokinetics.
- For modified-release products for which the fluctuation in plasma concentration over a dosage interval at steady state needs to be assessed.
- If problems of sensitivity preclude sufficiently precise plasma concentration measurements after single-dose administration.
- If the intra-individual variability in the plasma concentration or disposition precludes the possibility of demonstrating BE in a reasonably sized single-dose study and this variability is reduced at steady state.
- When a single-dose study cannot be conducted in healthy volunteers due to tolerability reasons and a single-dose study is not feasible in patients.
- If the medicine has a long terminal elimination half-life and blood concentrations after a single dose cannot be followed for a sufficient time.
- For those medicines that induce their own metabolism or show large intra-individual variability.
- For combination products for which the ratio of plasma concentration of the individual substances is important.
- If the medicine is likely to accumulate in the body.
- For enteric coated preparations in which the coating is innovative.
Under normal circumstances, blood should be the biological fluid sampled to measure drug concentrations. Most drugs may be measured in serum or plasma; however, in some drugs, whole blood (e.g., tacrolimus) may be more appropriate for analysis. If the blood concentrations are too minute to be detected and a substantial amount (40%) of the drug is eliminated unchanged in the urine, the urine may serve as the biological fluid to be sampled (e.g., alendronic acid).
Pharmacodynamic endpoint studies
Pharmacokinetic studies measure systemic exposure but are generally inappropriate to document local delivery BA and BE. In such cases, BA may be measured, and BE may be established, based on a pharmacodynamic study, providing an appropriate pharmacodynamic endpoint is available. Pharmacodynamic evaluation is measurement of the effect on a pathophysiological process, such as a function of time, after administration of two different products to serve as a basis for BE assessment. Regulatory authorities request justification from the applicant for the use of pharmacodynamic effects/parameters for the establishment of BE criteria. These studies generally become necessary fewer than two conditions 1) If the drug and/or metabolite(s) in plasma or urine cannot be analyzed quantitatively with sufficient accuracy and sensitivity; 2) If drug concentration measurement cannot be used as surrogate endpoints for the demonstration of efficacy and safety of the particular pharmaceutical product. The other important specifications for pharmacodynamic studies include:?
- A dose-response relationship should be demonstrated;
- Sufficient measurements should be taken to provide an appropriate pharmacodynamic response profile;
- The complete dose-effect curve should remain below the maximum physiological response;
- All pharmacodynamic measurements/methods should be validated for specificity, accuracy, and reproducibility. Examples of these pharmacodynamic studies include locally acting drug products and oral inhalation drug products, such as metered dose inhalers and dry powder inhalers, and topically applied dermatologic drug products, such as creams and ointments.
Bronchodilator drug products, such as albuterol metered dose inhalers, produce relaxation of smooth muscle of the airways. For these drug products, a pharmacodynamic endpoint, based either on increase in forced expiratory volume in 1 second (FEV1) or on measurement of PD20 or PC20 (the dose or concentration, respectively, of a challenge agent) is clinically relevant and may be used for BA and BE studies.
Clinical endpoint studies or comparative clinical trials
In the absence of pharmacokinetic and pharmacodynamic approaches, adequate and well-controlled clinical trials may be used to establish BA/BE. Several international regulatory authorities provide general information about the conduct of clinical studies to establish BE.
In vitro endpoint studies
More recently, a Biopharmaceutics Classification System (BCS) has categorized drug substances as having either high or low solubility or permeability and drug products as exhibiting rapid dissolution. According to this approach, drug substances may be classified into four primary groups:
1) Highly soluble and highly permeable;
2) Highly permeable and poorly soluble;
3) Highly soluble and poorly permeable;
4) Poorly soluble and poorly permeable.
Using this BCS approach, a highly permeable, highly soluble drug substance formulated into a rapidly dissolving drug product may need only in vitro dissolution studies to establish BE. In addition, in vitro approaches to document BE for nonbioproblem drugs approved before 1962 remain acceptable as per FDA regulations. Dissolution tests can also be used to reduce the number of in vivo studies in other circumstances, and to i) Assesses batch-to-batch quality and support batch release; ii) Provide process control and quality assurance; and iii) Assess the need for further BE studies relative to minor post-approval changes, where they function as a signal of bioinequivalence. The broad spectrum of BA/BE in vitro studies specifications were provided by each regulatory authority. 9-18
Statistical Analysis of Bioequivalence:
In the analysis of a bioequivalence study, the measurements of both Cmax and AUC are subject to the following procedure. The measurement for each subject is log transformed and the averages, µT and µR, of the test and reference products are calculated. The within subject variability of the reference product, σ 2 WR, is also calculated. There are two parts to the proposed bioequivalence criteria, a scaled average bioequivalence evaluation and a point estimate constraint. In order to demonstrate bioequivalence both parts must pass. Scaled average bioequivalence for both AUC and Cmax is evaluated by testing the following null hypothesis H0: [(µT- µR) 2 / σ2 WR] > ? (For given ? > 0) versus the alternative hypothesis H1: [(µT- µR) 2 / σ2 WR] ≤? where µT and µR are the averages of the logtransformed measure (Cmax, AUC ) for the test and reference products, respectively; usually testing is done at level α=0.05; and ? is the scaled average BE limit. Furthermore, ?= (ln?) 2 / σ2 Wo Where ? is 1.25, the usual average BE upper limit for the untransformed test/reference ratio of geometric means, and σ2 Wo =0.25. Note that rejection of the null hypothesis H0 supports the conclusion of equivalence. A 95% upper confidence bound for [(µT- µR) 2 / σ 2 WR] determined in a BE study must be ≤? or equivalently, a 95% upper confidence bound for (µT- µR) 2 / ?σ2 WR must be ≤0. Additionally, the point estimate (test/reference geometric mean ratio) must fall within [0.80, 1.25]. The test drug must pass both conditions before it is judged bioequivalent to the reference product. 19
HIGHLY VARIABLE DRUGS AND DRUG PRODUCTS
In bioequivalence evaluation, highly variable drugs are generally defined in the context of within-subject variability in bioequivalence parameters Cmax and AUC. The most oftenused definition of a highly variable drug is a drug which has a within-subject (synonymous with ?intra-subject?) variability of 30% or more in these two bioequivalence parameters. FDA‘s Office of Generic Drugs (OGD) estimates that approximately 10% of the submitted BE studies from Abbreviated New Drug Applications (ANDAs) showed some evidence of high variability. Examples exist where a highly variable reference product failed to demonstrate BE when compared to itself in a BE study using the standard design/sample size. As illustrated in Figure 2, because of this high variability, larger numbers of subjects may be needed in bioequivalence studies to give adequate statistical power to meet FDA bioequivalence limits. The FDA is currently investigating bioequivalence study design proposals that can reduce the number of subjects needed for a bioequivalence study.19-20 HVDs show variable pharmacokinetics as a result of their inherent properties (e.g. distribution, systemic metabolism and elimination). A drug may have low variability if it is administered intravenously, whereas it can be highly variable after oral administration. In such cases, the source of the high variability can be any of the processes that are involved in the absorption, such as problematic solubility, gastrointestinal instability, active transport or first-pass metabolism in the gut or liver. Davit et al. recently reviewed 1010 bioequivalence studies of 180 drugs, of which 31% (57 of 180) were highly variable.13 About 60% of the surveyed drugs were highly variable as a result of the pharmacokinetic characteristics of the drug substances. Several physicochemical and pharmacokinetic factors were considered that can potentially contribute to the observed high variation, such as low aqueous solubility, acid liability, low bioavailability (F), pronounced food effect and so on. Analysis of the data revealed that extensive first-pass metabolism was probably the most important factor. Eighty-three percent of the HVDs were subject to extensive first-pass metabolism, whereas the corresponding proportion in the non-highly variable group was 21%. In addition, the variability may be caused by the pharmaceutical form in which the drug is contained. In this case, different formulations of the same drug may show different within-subject variabilities (e.g. nadolol). The distinction between HVDs and HVDPs is especially important with modified release dosage forms and in formulations of poorly soluble drugs (Biopharmaceutics Classification System classes II and IV), where the formulation factors are more important. Davit et al. related the variabilities of dissolution performance to those of bioequivalence parameters. The results suggested that in about 20% of the highly variable cases, the performance of drug formulations could contribute to the high variation. 13, 20
The factors described above influence bioequivalence parameter variability due to the characteristics of the drug substance, rather than those of the drug product. Drug product formulation can also contribute to high variability in bioequivalence parameters. For example, if the rate of drug release from the dosage form is highly variable, this factor may cause high variability in bioequivalence parameters and may signify a product with lower product quality. Figure 3 diagrams the steps involved in bioequivalence evaluation of oral dosage form performance and illustrate ways in which high within-subject variability in bioequivalence measures can arise from either the drug substance or the drug product.13
Identification of Highly Variable Drugs
The RMSE (Root Mean Square Error) values of the bioequivalence parameters Cmax and AUC0-t was used as an estimate of within-subject variability. Since most of the studies submitted to the DBE (Division of Bioequivalence) used a two-way crossover design; it was not possible to determine the true within-subject variability. Therefore, the RMSE was used as an estimate of within-subject variability. Since highly variable drugs are defined as drugs with within subject variability of 30% or more in bioequivalence parameters, we considered a drug to have high within-subject variability if the RMSE for either AUC0-t or Cmax was ≥0.3.
Although the FDA evaluates AUC∞ in bioequivalence studies, but did not define a highly variable drug as one for which the AUC∞ RMSE≥0.3 because the calculations necessary to extrapolate to infinity contribute to the variability of this measure. Therefore, we consider AUC0-t to be a better indicator of variability due to drug substance and/or drug product than AUC∞. Table 2 shows the number of bioequivalence studies, drug products, and drugs reviewed by the DBE in 2003–2005. During this time period, the DBE found acceptable 1,010 bioequivalence studies. These 1,010 bioequivalence studies investigated a total of 524 different drug products, for 180 different drugs. Frequently, there are at least several generic versions of any one reference listed drug under review at the OGD during the same time period. Each new generic drug product line is usually the subject of a separate ANDA. Most ANDAs contain at least two bioequivalence studies, one under fasting conditions and one under fed conditions. A minority of ANDAs contains either one fasting bioequivalence study or one fed bioequivalence study. In 111 of these 1,010 acceptable studies, the RMSE was ≥0.3 for either Cmax and/or AUC0-t. As our criteria for classification as a highly variable drug was that the RMSE≥ 0.3 for Cmax and/or AUC0-t, we concluded that 111 or 11% of these studies were of drug products that showed high variability in bioequivalence parameters. These 111 studies of highly variable drugs were of 101 different drug products, representing 57 different drugs. 13 Determination of whether high variability in bioequivalence parameters was consistent We further classified drugs for which the RMSE for Cmax and/or AUC0-t≥0.3 as consistently highly variable, borderline highly variable, or inconsistently highly variable. Table 3 was subject to extensive first pass metabolism). As these are properties of, or factors influencing, the disposition of the drug substance, we concluded that 61% of the highly variable drugs reviewed in 2003–2005 were likely highly variable due to drug substance characteristics. Notably, several drugs in each of the following classes were in the consistent and borderline highly variable groups: angiotensin converting enzyme (ACE) inhibitors, calcium channel blockers, 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) inhibitors, and bisphosphonates. All of the ACE inhibitors reviewed during the 2003–2005 period are inactive prodrugs that undergo extensive first-pass metabolism. The calcium channel blockers and HMG-CoA inhibitors reviewed during this period are also known to undergo extensive first pass metabolism. The bisphosphonate drugs reviewed during this period are reported to have absolute oral bioavailability averaging less than 1%. Thus, for some potential generic drug products, it may be possible to predict whether variability in bioequivalence parameters will be high based on what is known about the physicochemical and dispositional characteristics of the drug class in general. 13
Significance of the HVD Problem;
Although the problem is well known, it is still very difficult to get hard figures about its extent. An overview of FDA submissions showed that about 15% of the applications fell into the category of HVDs. At first sight, the issue of HVDs did not appear to be so serious, because all submitted applications for HVDs passed the 0.80–1.25 regulatory criterion. However, this regulatory experience was not shared by other parties involved in generic drug development. An overview of the database of a well known Canadian contract research organization showed a considerably different picture. In a review of 580 studies, 105 fell into the highly variable category. The failure rate was 54%. A very similar figure was reported by another large Canadian contract research organization. It appears that only bioequivalence studies meeting the regulatory expectations were submitted by applicants and, consequently, the regulatory agencies could have underestimated the seriousness of the HVD problem. Some studies are not undertaken at all when the very heavy financial and logistical difficulties are confronted. Diliberti presented a case where the within subject variation was 173%for the Cmax and 157%for the AUC at time t (AUCt). He estimated that at least 598 subjects would be needed to meet the 0.80–1.25 criterion with 80% power. The issue of bioequivalence for hvds is not only a problem for the generic industry It has been implicitly assumed until this point that the single role of bioequivalence studies is to gain marketing authorization for generic products. That is not so. For example, it is very common that a drug formulation used in early clinical studies is different from that applied in the late, pivotal investigations. In this case, the innovator company performs a ?bridging‘ bioequivalence study in order to demonstrate that the formulation change does not have a clinically significant impact. These studies are typically powered to meet the 0.80–1.25 bioequivalence criterion, partly because of the convention and partly because at that stage of product development, there are no firm clinical safety data. Also, following Hauschke et al. and Steinijans et al. the paradigm of bioequivalence is used to evaluate the drugdrug and drug-food interactions. In the case of drug interactions, the lower and upper limits of the bioequivalence ranges could be different from 0.80 and 1.25, and alternative ?effect boundaries‘ could be allowed on the basis of concentration response relationships, pharmacokineticpharmacodynamic models or other available information. For example, a lack of a food effect is not considered to be established if the CI is outside the 0.80–1.25 limits. Altogether, if a new drug has highly variable features, then to establish bioequivalence between formulations used in the product development process or to demonstrate dose linearity can be a difficult and expensive challenge. For these reasons, HVDs are not just a problem for the generic industry but are also a source of substantial concern to the innovators. However, compared with generic producers, regulatory agencies are rather tolerant to the innovators‘ request for post hoc widening based on clinical grounds. Because of this, concerns of innovators about large sample sizes are much less apparent. 21
Proposals from the Literature As indicated, the bioequivalence criteria in the U.S. recommend that the 90% confidence interval of the geometric mean ratio between the test and reference products fall within 80-125%. Over the years, various suggestions have been made in an attempt to alleviate the difficulty of meeting the bioequivalence limits for highly variable drugs and drug products. Various authors have explored the use of replicate designs or group-sequential designs. If a subject-by-formulation interaction is negligible, the sample size required for a replicate design study can be reduced up to 50% of that for a non-replicate design study the number of study periods is the same since approximately half the usual number of subjects is used but they are each studied for twice as many periods. Therefore, it takes a longer time to complete a replicate design study, resulting in an increased chance of subject dropout from the trial. A group-sequential design may be useful in cases where there is uncertainty about the estimates of variability. Nonetheless, the total number of subjects employed with this design may be the same as that used for a study without the group-sequential design if the interim analysis does not indicate bioequivalence. Also, to preserve the overall Type I error rate of 5%, a higher level of confidence interval has to be used at each stage of the interim analysis.22 Several proposals are available in the literature to modify the existing bioequivalence criteria for highly variable drugs and drug products. In general, these various criteria are based on either the reduction of the level of the confidence interval or an increase of the width of the equivalence limits, or both. The level of confidence interval reflects the degree of consumer risk (Type I error in statistical terms) that can be tolerated by the regulatory agencies. A reduction in the level of confidence interval, for example, from 90% to 85%, implies a possible increase in the consumer risk, which would not be in the best interests of public health. In contrast, the width of equivalence limits represents the allowable boundary for the ratio (or difference) of the means between products in comparison. Any adjustment of these limits should be based on consideration of the statistical properties of the data as well as on the clinical characteristics of the individual drug. Statistically, widening the bioequivalence limits can be accomplished through expansion of the allowable boundary or by scaling the criteria based on the high variability of the reference product. 23-24
PROPOSED SOLUTIONS FOR THE PROBLEM OF HVD
Relaxation of the Regulatory Requirement: Health Canada generally expects only that the point estimate of the GMR (Geometric Mean Ratio) for the Cmax, but not its 90% CI, should be between the regulatory limits of 0.80 and 1.25. This relaxed requirement applies generally and is not aimed specifically at HVDs. Nevertheless, it enables satisfactory determination of bioequivalence for several HVDs because the variation of the Cmax is usually higher than that of the AUC, and therefore the determination of bioequivalence can often fail because of the wide CI of the Cmax. 21 Widening of Bioequivalence Limits Based on Reference Variability The bioequivalence limits for these methods are not determined by the sample size. Rather, they will be scaled based on the within-subject variability of the reference product. For both Methods 2 and 3 below, a side condition to constrain the mean difference between the test and reference products has also been proposed
Method 1:
The rationale for this approach is that a mean difference of 25% is considered small relative to the range of values an individual may experience when the within-subject variability is high, e.g., 40%. Therefore, the acceptable limits may be scaled in relation to the size of within-subject variability as follows: [U, L] = Exp [σWR] Where U and L are the upper and lower limits, respectively; k represents the pth percentile of the standard normal distribution, Zp; and WR is the estimated within-subject standard deviation (obtained from the ANOVA on the log scale) for the reference. When k = 1, ~ 67% of the pharmacokinetic measures (such as AUC) experienced by an individual will be within the range of [U, L]. Table 4 lists the choices of limits at k = 1.
Method 2:
A scaled average bioequivalence criterion has been proposed [(µT- µR) 2 / σ 2 WR] ≤? Where µT and µR are the averages of the logtransformed measure for the test and reference products, respectively; and ? is the bioequivalence limit. Comparing Methods 1 and 2, it can be seen that k = ? -1/2 = (ln1.25)/ σ2 Wo where W0 is the cutoff within-subject standard deviation for scaling. Relationship of k and σW0 are given in Table 5.
Method 3:
Derived from the comparison of the distance measure between the test and reference products, the following individual bioequivalence criterion has a reference variance in the denominator, and thus is scaled to the reference variability. [(µT- µR) 2 + (σ2 WT- σ 2 WR) + σ2D]/ σ2 WR ≤? Where WT is the estimated within-subject standard deviation for the test product; 2D is the subject-by-formulation interaction variance component; and I is individual bioequivalence limit. Although theoretically sound, the individual bioequivalence criterion requires replicate designs and inclusion of target population in the study. Because of these resource implications, the FDA has recommended the continued use of an average criterion to compare bioavailability measures.22-27
Direct Expansion of Bioequivalence Limits Sample size in bioequivalence studies is determined in large part by the bioavailability parameter with the highest variability. In most cases, Cmax has higher variability than AUC. Thus, widening of the bioequivalence limits for Cmax has been proposed to reduce the sample size needed in the evaluation of bioequivalence for highly variable drugs/products. The greater variability observed with Cmax may result from the fact that this parameter is a single point measurement, which is highly dependent on the sampling time/frequency and elimination rate of the drug. The EMEA currently allows for expanded limits (e.g., 69.84-143.19%) for Cmax in certain cases where no safety or efficacy concern arises, based on the consideration of higher variability for this measure as compared to AUC.15 Expansion of Bioequivalence Limits Based on Fixed Sample Size This method was proposed based on the notion that only a reasonable number of subjects should be required for a bioequivalence study.23, 28 The number of subjects is fixed by a standard two-period, crossover study comparing the reference product with itself where the study fails to meet the 80-125% limit. The confidence interval obtained from the reference product in this study would become the ?goalposts? for the subsequent studies comparing the test with reference product, using the same number of subjects. Expansion of Bioequivalence Limits Based on Sample Size and Scaling In addition to fixing the sample size, this method takes into consideration the producer‘s risk (Type II error) and reference variability.23 the equation for the allowable limits is: [U, L] = Exp [± (tα + tβ/2) n -1/2 σ WR] ……. (Eq.1) Where α and β are the consumer and producer risks, respectively; 2n is the number of subjects desired in the study; and t is the percentile of the t-distribution with 2n-2 degrees of freedom. The current regulatory standard has kept the consumer risk at a level of no more than 5% while allowing the drug applicant or sponsor to control its own producer risk. Based on Eq. 1, for example, assuming a 5% consumer risk and 10% producer risk, the proposed bioequivalence limits for a typical sample size of 24 subjects will be (0.74, 1.35) at σ WR = 0.3 (0.61, 1.65) at σ WR = 0.5 Recent Considerations by Regulatory Agencies Although global harmonization is a general goal, to date, bioequivalence has not been accepted as a topic by the International Conference on Harmonization (ICH). Nonetheless, the resource and ethical concerns for highly variable drugs/products in bioequivalence are generally recognized by international regulatory agencies. It is thus useful to review the differing regulatory approaches before an informed recommendation is made on the topic. The following outlines the bioequivalence standards used in different regions: In Canada, for drugs with uncomplicated characteristics, a 90% confidence limit of 80- 125% is required for AUC. However, a limit is placed only on the means (or point estimate) for Cmax) 11. As a result of random variation or a larger than expected relative difference, the sponsor may add more subjects. If this option is chosen, it must be stated in the study protocol. In addition, two criteria must be met before combining is acceptable: 1) The same protocol must be used; and 2) Consistency tests must be met at an alpha error rate of five percent. The European Agency for the Evaluation of Medicinal Products (EMEA) has similar bioequivalence standards to those in the FDA, i.e., 90% confidence limits of 80-125% on AUC and Cmax, with the qualification that these limits may be expanded in certain cases for Cmax (e.g., 69.84-143.19%) provided that there is no safety or efficacy concerns.15 In Japan, the bioequivalence standards also rely on the 90% confidence limits of 80-125% for both AUC and Cmax, although wider limits are allowed for less potent drugs. Additionally, if the confidence limits are outside of 80-125%, bioequivalence may be claimed on the grounds that the study meets. 10 All three conditions listed below: 1) The total number of subjects in the initial bioequivalence study is no less than 20 (n=10/group), or pooled sample size of the initial, 2) The differences in average values of logarithmic AUC and Cmax between two products are between log (0.9) – log (1.11); and 3) Dissolution rates of test and reference products are determined to be equivalent under all dissolution testing conditions specified. Japan allows the addition of subjects to increase the power of a failed bioequivalence study. However, the add-on subjects cannot be less than half the number in the original study. South Africa accepts an acceptance interval of 75-133% for Cmax, except for narrow therapeutic range drugs, when an acceptance interval of 80-125% applies. For highly variable drugs, a wider interval or other appropriate measure may be acceptable, but should be stated a priori and justified in the protocol.25
Evaluation of Bioequivalence with SABE
Regulatory authorities appear to move towards adopting the approach of scaled average bioequivalence (SABE) as a tool for dealing with the problem of bioequivalence for HV drugs. Therefore, a brief background of the procedure will be summarized. The two one-sided tests procedure is generally applied for determinations of bioequivalence. In practice, BE is evaluated by calculating logarithmic quantities. Thus, means and standard deviations of the logarithmic data (µ and σ) are estimated. Bioequivalence is declared if the difference between the logarithmic averages is between limits (BELA) which are preset by regulatory authorities. Therefore, average bioequivalence (ABE) is accepted if the following criterion is satisfied:
- BELA ≤ μT - μR ≤ BELA The most usually applied regulatory limit is: BELA = ln (1.25) (1A) This assures the earlier stated expectation that the regulatory limits for the ratio of geometric means of metrics are 0.80 and 1.25. In practice, the 90% confidence interval around the difference between the estimated logarithmic averages should be between the regulatory limits. Thus, regulators need to define, in the case of average BE, a single criterion for declaring bioequivalence such as that given in Eq. (1A). For highly-variable drugs, evaluated by scaled average BE, two quantities must be defined. They will be discussed below: The regulatory criterion suggested for the application of scaled average BE is: -BELS ≤ (μT-μR)/σW ≤ BELS (2) Here a scaling standard deviation (σW) is related to the within-subject standard deviation of the reference formulation (σWR) or, in other views, is identical to it. This distinction will be discussed later. Tothfalusi et al. suggested that the scaled BE limits (BELS) should be set in the following form: BELS = ln (1.25)/σ0 (2A) Here σ0 is the first measure which should be defined by regulators. It will be referred to as the regulatory standardized variation. It defines the proportionality factor between the logarithmic BE limits and σW in the highly-variable region (see Figure 4A). σ0 uniquely determines BELs and vice versa. For example, when σ0 = 0.294 then BELs is 0.759, and when σ0 = 0.246 then BELs is 0.907. Rearranging equation (2), an alternative form is obtained: -BELs σW ≤ μT-μR ≤ BELs σ This form represents average bioequivalence with expanding limits (ABEL). Consequently, Eq. 2 and Eq. 2B, i.e. the approaches of SABE and ABEL, are (almost) identical. Using the limits of ABEL helps to understand the properties of SABE from the perspective of ABE. In this context, the regulatory standardized variation (σ0) defines the proportionality factor between the logarithmic ABEL limits and σW (Figure 1A). A representation of ABEL conveniently illustrates a mixed regulatory strategy that was proposed for applying the unscaled and scaled approaches to the determination of BE (Figure 4). According to the mixed regulatory strategy, a second regulatory term, the so-called switching variation (CVS), separates regions of low and high variabilities. If the variation of the drug is low, i.e., when it does not exceed the switching variation (CVW ≤ CVS) then, following the present practice, unscaled average BE should be evaluated. However, for HV drugs when the variability is higher than the switching variation (CVW > CVS), scaled average BE is applied. The mixed regulatory strategy is depicted in Figure 4 where, for illustrative purposes, SABE equivalent ABEL limits (BELE* σ ) are plotted. Two different SABE-equivalent ABEL limits are shown which correspond to two different values of σ0. How to set σ0 is the main focus of this communication. The standard deviations (σ) can be converted, approximately, to the corresponding coefficients of variation: CV = 100[exp (σ2 )-1)] 1/2 (3) Therefore, for unified and convenient treatment, the regulatory constants are expressed in terms of coefficients of variation. As an alternative notation, CV0 will be used instead of σ0 and the transformation rule between CV0 and σ0, given by Eq. 3, will be applied. For example, if σ0 = 0.294 then CV0 = 30%, and when σ0 = 0.246 then CV0 = 25%. The advantage of this unified notation is that an additional GMR restriction rule also can be expressed in relative terms. The 0.80-1.25 GMR restriction criterion becomes a regulatory constraint of 25%. Thus, in our notation, the proposed mixed approach depends on three regulatory constants, CVs, CV0 and CVGMR, with typical values of 30%, 30% and 25%.19-29 Considerations on the implementation of scaled average bioequivalence: the recommendations of FDA As noted earlier, the Advisory Committee for Pharmaceutical Sciences discussed the topic repeatedly. At its meeting, on October 6, 2006, important presentations were offered on behalf of the FDA Working Group on Highly Variable Drugs (16-18). The interim recommendations of FDA were further clarified on May 22, 2007 at an AAPS/FDA workshop. The current proposals of FDA and their quantitative characteristics were published very recently. FDA has proposed to apply the approach of reference-scaled average BE for determining the BE of HV drugs. This means that σW = σWR would be adopted for scaling. FDA suggests also that the acceptance criteria include a constraint on the point estimate for the ratio of geometric means (GMR). It recommends that GMR be limited to the range of 0.80 to 1.25. The Advisory Committee concurred with this proposal but some members actually favoured a narrower range. FDA proposes that both AUC and Cmax should satisfy the BE acceptance criteria. FDA recommends that three-period BE studies be performed in which the reference product (R] is provided twice and the test product (T) is given once. Consequently, the possible sequences of drug administration are TRR, RRT, and RTR. The FDA Working Group performed simulations in order to ascertain the features of the above proposals. The current FDA recommendations include a value of σ0 = 0.25. FDA suggests also that unscaled average BE used if the within-subject variability is less than 30%, and that reference-scaled average BE applied if the within-subject variability is at least 30%. These suggestions correspond to a switching coefficient of variation of CVS = 30%.19-29 Proposed Study Design For drugs with an expected within-subject variability of 30% or greater, a BE study with three-period, reference- replicated, crossover design with sequences of TRR, RTR, and RRT is proposed. Specifically, subjects receive a single dose of the test product once and reference product twice on separate occasions with random assignment to the three possible sequences of product administration. This partial replicate design allows for the estimation of within subject variability for the reference product. Treatments should be separated by a washout period of adequate duration such that the drug of interest can no longer be detected in plasma. Subjects recruited for in vivo BE studies should be 18 years of age or older and capable of giving informed consent unless otherwise indicated by a specific guidance. It is the sponsor‘s responsibility to determine the sample size needed to achieve the desired power in a study; however, the minimum number of subjects that would be acceptable is 24. The three-period design was selected over a four-period design because of efficiency. The only advantage of the four period designs is that it allows the calculation of the variability of the test product. The test product variability is not used in the proposed statistical method. Some concern has been raised that an ANDA sponsor may produce a product that has higher variability than the reference product. However, under the recommended design, ANDA sponsors that design a product of lower variability than the reference product will need a smaller number of subjects to pass. A disadvantage of the four-period design is that the dropout rate for studies increases with the length of the study. Nevertheless, sponsors may use the four-period design to demonstrate the BE for their highly variable drug products.
DISCUSSION AND CONCLUSION
The impact of Cmax variability on the determination of bioequivalence, as well as the possible approaches to resolving this issue has been discussed extensively in the published literature. Major regulatory agencies have provisions in their regulations which can accommodate the effect of higher variability associated with cmax on the design of bioequivalence studies. For example, health canada does not require any limits on the confidence interval for cmax, although limits are placed on the point estimates for this parameter. the EMEA and Medicines Control Council of South Africa both allow for expanded limits for cmax in certain cases provided that there are no safeties or efficacy concerns. Similarly, the Japanese division of drugs accepts limits greater than 80 – 125%, ?for drugs with pharmacologically mild actions?. Additionally, a failed bioequivalence study can utilize additional subjects to increase power and the likelihood of meeting be criteria, provided other conditions are met. This report presents a proposal for the BE evaluation of highly variable drugs and drug products. This new approach addresses many of the concerns about the BE of highly variable drugs/products that have been raised for the past several years. The proposed approach adjusts the BE limits of highly variable drugs/products by scaling to the within subject variability of the reference product in the study. The recommendation for the use of reference-scaling is based on the general concept that reference variability should be used as an index for setting the public standard expressed in the BE limit. Furthermore, for drugs and products that are highly variable, reference-scaling effectively decreases the sample size needed for demonstrating BE. The additional requirement of a point-estimate constraint will impose a limit on the difference between the test and reference means, thereby eliminating the potential that a test product would enter the market based on a bioequivalence study with a large mean difference. The use of the reference-scaling approach necessitates a study design that evaluates the reference variability, via multiple administration of the reference treatment to each subject. The recommended 3-period design is the most efficient way to obtain this information. The proposed approach will resolve a number of issues in the BE evaluation of highly variable drugs while achieving the FDA‘s mission of ensuring that all the drugs approved for use in U.S. are both safe and effective.
ACKNOWLEDGMENTS
I am indebted to my esteemed guide Dr. Anil Bhandari (Dean, Faculty of Pharmaceutical Sciences, Jodhpur National University) for his excellent guidance, export suggestions, encouragement, support, lively discussion, constructive criticism, insightful corrections and everlasting interest in pharmacology and.
References:
1. Asif M. Tamboli, Pavan Todkar, Priti Zope, Sayyad FJ. An Overview on Bioequivalence: Regulatory Consideration for Generic Drug Products. J Bioequiv Availab 2010; 2(4): 086-092.
2. Biomarkers and Surrogate Endpoints: Advancing Clinical Research and Applications. National Institutes of Health and the Food and Drug Administration Conference. April 15–16, 1999, Bethesda, Maryland.
3. US Code of Federal Regulations Title 21, Section 320.1(e). US Government Printing Office, Washington, DC, 2006.
4. US Food and Drug Administration. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Considerations, March 2003.
5. Patterson SD, Zariffa NMD, Montague TH, Howland K. Non-traditional study designs to demonstrate average bioequivalence for highly variable drug products. Eur J Pharm Sci 2001; 57:663-670
6. Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm 1987; 15:657-680.
7. U. S. Food and Drug Administration. Guidance for Industry: Statistical Approaches to Establishing Bioequivalence, January 2001.
8. FDA Orange Book, Preface p. viii. http://www.fda.gov/cder/ob/ default.htm. Accessed on dated Nov. 8, 2011.
9. Therapeutic Goods Administration. CPMP Guideline "As adapted in Australia by the TGA" With Amendment - Note for Guidance on the Investigation of Bioavailability and Bioequivalence (CPMP/EWP/QWP/1401/98). Effective 10 April 2002. http:// www.tga.gov.au/docs/pdf/euguide/ewp/1401 98entga.pdf. Accessed on dated Nov. 8, 2011.
10. Guideline for Bioequivalence Studies of Generic Products (December, 2006). National Institute of Health Sciences. Japan NIHS. http://www. nihs.go.jp/drug/be-guide (e)/be2006e.pdf. Accessed on dated Nov. 8, 2011.
11. Canadian Health Protection Branch (HPB), Health Canada. Guidance for Industry Conduct and Analysis of Bioavailability and Bioequivalence Studies – Part A: Oral Dosage Formulations Used for Systemic Effects. Published by authority of the Minister of Health. 1992. Health Products and Food Branch Guidance Document. http://www.hc-sc.gc.ca/dhpmps/alt_formats/hpfbdgpsa/pdf/prodpharma/bio-a-eng.pdf. Accessed on dated Nov. 8, 2011.
12. Asean Guidelines for the Conduct of Bioavailability and Bioequivalence Studies. http://www.suregmp.com/forum/file_down_ ok.asp?Folder=PDSandDownFile=Bioavailabi lty_Bioequivalence%20Stdies.pdf. Accessed on dated Nov. 8, 2011.
13. Davit BM, Conner DP, Fabian-Fritsch B, et al. Highly Variable Drugs: Observations from Bioequivalence Data Submitted to the FDA for New Generic Drug Applications. AAPS J 2008; 10(1):148-156.
14. Medicines Control Council. Biostudies. June 2007. http://www.mccza.com/generic Documents/2.06_Biostudies_Jun07_v2.zip. Accessed on dated Nov. 8, 2011.
15. European Medicines Agency. London, 24 May 2007. Doc. Ref. EMEA/ CHMP/EWP/200943/2007. Committee for Medicinal Products for Human Use (CHMP). Recommendation on the Need for Revision of (CHMP).Note For Guidance on the Investigation of Bioavailability and Bioequivalence. CPMP/EWP/QWP/1401/98. http://www.ema.europa.eu/docs/en_GB/doc ument_library/Scientific_guideline/2009/09/ WC500003009.pdf Accessed on dated Nov. 8, 2011.
16. European Medicines Agency. PreAuthorisation Evaluation of Medicines for Human Use London, 24 July 2008. Doc. Ref. CPMP/EWP/ QWP/1401/98 Rev. 1. Committee for Medicinal Products for Human Use (CHMP). Draft. Guideline on the Investigation of Bioequivalence. http://www.ema.europa.eu/docs/en_GB/doc ument_library/Scientific_guideline/2009/09/ WC500003011.pdf Accessed on dated Nov. 8, 2011.
17. Kingdom of Saudi Arabia Saudi Food and Drug Authority Drug Sector. Bioequivalence Requirements Guidelines. Draft. May 2005. http://www.sfda.gov.sa/NR/rdonlyres/6A11 4B70-4201-46EF-B4C7- 127FD66D3314/ 0/BioequivalenceRequirementGuidelines.pd f. Accessed on dated Nov. 8, 2011.
18. Guidance Document for Bioequivalence Study of Korea Food and Drug Administration Notification #2008–22 (May 07, 2008). http:// betest.kfda.go.kr/country/GUIDANCE%20F OR%20INDUSTRY%20 (KFDA_2005).pdf. Accessed on dated Nov. 8, 2011.
19. Haidar SH, Barbara D, Chen ML, Conner D. Bioequivalence Approaches for Highly Variable Drugs and Drug Products. Pharm Res 2008; 25(1):237-241.
20. Midha KK, Rawson MJ, Hubbard JW. The bioequivalence of highly variable drugs and drug products. Int J Clin Pharmacol Ther 2005; 43 (10): 485-98.
21. Tothfalusi L, Endrenyi L, Arieta AG. Evaluation of Bioequivalence for Highly Variable Drugs with Scaled Average Bioequivalence. Clin Pharmacokinet 2009; 48 (11): 725-743.
22. Patterson, SD, Zariffa NMD, Montague TH, Howland K. Non-traditional study designs to demonstrate average bioequivalence for highly variable drug products. Eur J Clin Pharmacol 2001; 57:663-670.
23. Boddy AW, Snikeris FC, Kringle RO, Wei GCG. Oppermann JA, Midha KK. An approach for widening the bioequivalence acceptance limits in the case of highly variable drugs. Pharm Res 1995; 12:1865- 1868.
24. Tothfalus L, Endrenyi, L, Midha, KK, Rawson MJ, Hubbard. JW. Evaluation of the bioequivalence of highly variable drugs and drug products. Pharm Res 2001; 18:728- 733.
25. Medicines Control Council, Department of Health, Republic of South Africa. Registration of Medicines: Biostudies. 2003
26. Tothfalus L, Endrenyi L, Midha KK. Scaling or wider bioequivalence limits for highly variable drugs and for the special case of Cmax. Int J Clin Pharmacol Ther 2003; 41:217-225.
27. Tothfalus L, Endrenyi L. Limits for the scaled average bioequivalence of highly variable drugs and drug products. Pharm Res 2003; 20:382-389.
28. Blume H, Midha, KK. Report of Consensus Meeting: Bio-international ‘92. Conference on Bioavailability, Bioequivalence and Pharmacokinetic Studies. Eur J Pharm Sci 1992; 1:165-171.
29. Endrenyi L, Tothfalusi L. Regulatory Conditions for the Determination of Bioequivalence of Highly Variable Drugs. J Pharm Pharmaceut Sci 2009; 12 (1): 138 – 149.
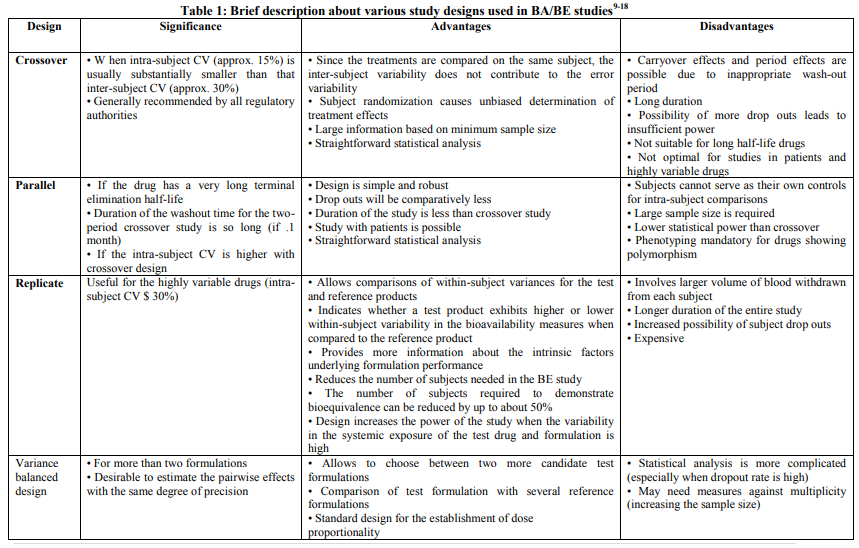
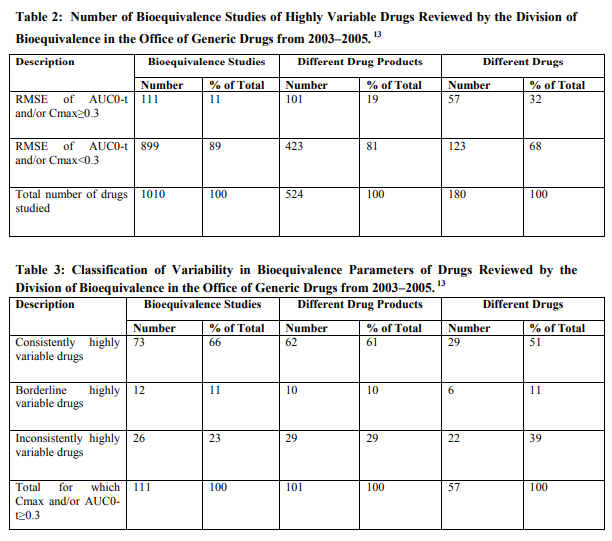
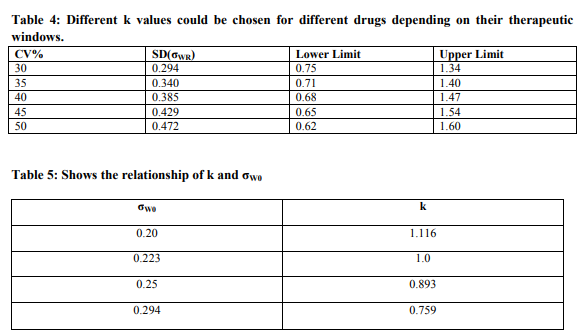
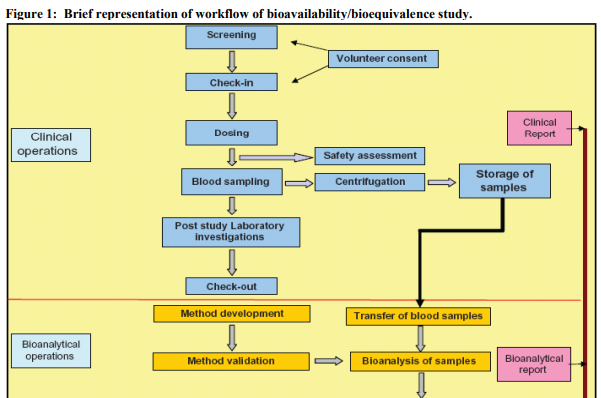
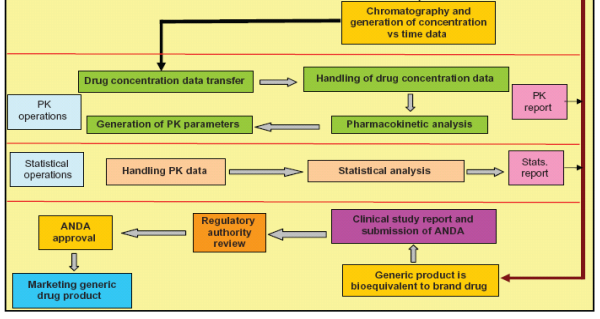
Figure 2: A visual representation of some possible results of the statistical analyses of bioequivalence studies. The three bars represent the widths of hypothetical 90% confidence intervals from bioequivalence studies of drugs with normal variability (green bar), low variability (blue bar), and high variability (red bar). A bell-shaped curve is superimposed over green bar, representing the 90% confidence interval, distributed around the geometric mean test/ reference ratio (?point estimate?), for the normal variability drug. For simplification, blue and red bars, respectively, are used in this diagram to represent confidence interval widths of low variability and highly variable drugs. The blue and red bars also actually represent the 90% confidence intervals of the bioequivalence study Cmax or AUC test/reference ratios normally distributed about the point estimate. The FDA concludes that a test and reference product are bioequivalent if the 90% confidence intervals (expressed as a percent) of the geometric mean Cmax and AUC test/reference ratios fall within the bioequivalence limits of 80–125%. In this illustration, the 90% confidence interval of the normal variability drug (green bar) meets bioequivalence limits. The 90% confidence interval of the drug with low variability meets bioequivalence limits although the point estimate deviates from 1.00. For a highly variable drug, the 90% confidence interval can exceed bioequivalence limits solely because of the variability. Using more subjects in the bioequivalence study will cause the 90% confidence interval of a highly variable drug to become narrower.20
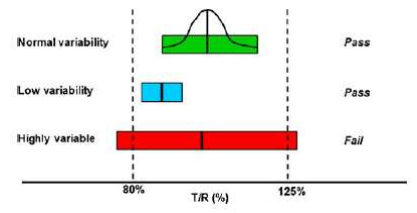
Figure 3: A diagram relating solid oral dosage form performance to the in vivo system in a bioequivalence study. Once ingested, a solid oral dosage form disintegrates, then dissolves into solution (formulation stage). The dissolved drug is absorbed through the gut wall, enters the liver through the portal vein, and from the liver goes into the systemic circulation, where pharmacokinetic measurement is possible. From the systemic circulation, the drug reaches the site of activity from which one observes a clinical response, where pharmacodynamic or therapeutic measurement is possible. Although the most accurate way of determining bioequivalence would be to compare test and reference product performance at the formulation stage, this is nearly always not possible. Consequently, most bioequivalence studies of systemically absorbed drugs rely on pharmacokinetic measures, as drug blood concentrations are thought to directly relate to the amount of drug released from the dosage form. Therefore, a properly designed in vivo study with pharmacokinetic endpoints can accurately determine whether a test and reference product is bioequivalent. As the drug moves from the formulation to the systemic circulation to the site of activity, the pharmacokinetic or pharmacodynamic response becomes increasingly variable with increasing numbers of steps between the formulation, pharmacokinetic measurement stage, and pharmacodynamic measurement stage. For example, for drugs that undergo extensive presystemic metabolism, the effects of the various biotransformation(s) brought about by various gut wall and/or hepatic metabolism steps contribute to the variability observed in drug pharmacodynamic measurements. This figure also illustrates the two sources of variability in bioequivalence measures—variability due to drug substance pharmacokinetics versus variability due to drug product performance. If high variability exists due to drug substance pharmacokinetics, it may be necessary to use large numbers of subjects to achieve an acceptable bioequivalence study. However, if the high variability is due to the formulation or dosage form performance, this may reflect either a poor quality test or reference product.13
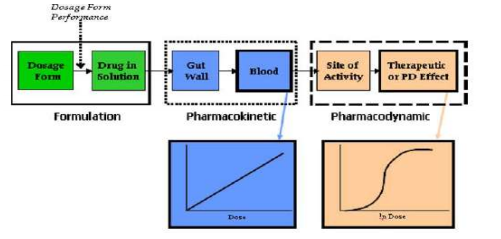
Figure 4: Mixed regul atory model for the determination of bioequivalence. The logarithmic BE limits, for determinations of average BE with constant and expanding limits, are shown by thick lines. If the within-subject variation (CVW) does not exceed the switching variation (CVS) then unscaled average BE is applied, and the BE limits have a constant level of ±log(1.25). When the within-subject variation is higher than the switching variations then the limits widen with increasing within-subject variation, and scaled average BE can be applied. The slope (in the logarithmic scale) of the expansion is determined by the regulatory standardized variation (CV0). The logarithmic average and the SABE-equivalent BE limits are shown by thick lines. (A) The regulatory standardized variation equals the switching variation, CV0=CVS=30% (B) the regulatory standardized variation is lower than the switching variation, CV0 = 25% and CVS = 30%. The BE limits have a discontinuity at the switching variation.19-29
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





