IJCRR - 10(13), July, 2018
Pages: 05-10
Date of Publication: 10-Jul-2018
Print Article
Download XML Download PDF
Validated RP-HPLC Method for Quantification of Paclitaxel in Human Plasma \? Eliminates Negative Influence of Cremophor El
Author: Hindu Kalluru, Vinodhini C., Satish Srinivas K., Surulivel Rajan M., Chitra K., Mangathayaru K.
Category: Healthcare
Abstract:Background: Literature reports innumerable methods for quantification of paclitaxel in biological matrices. Most of these involve complicated extraction procedures like solid phase extraction, separate procedure for elimination of interference of Cremophor El, advanced and expensive instruments.
Objectives: The objective of the present research work is to develop and validate a simple, rapid, sensitive, economic and reproducible reverse phase \? high performance liquid chromatography method for the estimation of paclitaxel concentration in human plasma that eliminates negative influence of Cremophor El on recovery of paclitaxel.
Methods: Chromatographic separation of paclitaxel was carried out using C18 column (150 × 4.6 mm i.d., 4\?m particle size, Waters, Australia) with 60% acetonitrile, 40% of 10mM ammonium acetate buffer solution and 0.1% formic acid as a mobile phase at a flow rate of 1.0mL/min at ambient temperature. Validation was performed as per ICH Q2 guidelines.
Results: In this system the retention time was 3min. The detection limit was 5ng/mL and limit of quantification with reproducibility was 15ng/mL. Plasma samples were extracted using single solvent (tertiary \? Butyl Methyl Ether) liquid- liquid extraction with a recovery of 96-99%. Robustness of the method was established with variation in flow rate, detection wave length and mobile phase composition. Stability of paclitaxel during the study period was studied and found to be stable.
Conclusion: The developed method is easy to perform, quick, reproducible with good recovery without negative influence of Cremophor El and applicable to quantify paclitaxel in regular clinical practice for individualization of the therapies.
Keywords: Paclitaxel, Cremophor El, Single solvent extraction, Bioanalytical
Full Text:
Paclitaxel is a very effective antineoplastic agent used in the treatment of ovarian, breast, cervix and non-small cell lung cancers (1). Paclitaxel was the first taxane derivative isolated from the bark of Taxusbrevifoliain the year 1971 (2) and being marketed from 1993 (3). It is a complex, hydrophobic molecule administered in a solution of Cremophor-EL (Cr El) and dehydrated alcohol in the ratio of 1:1.
Neutropenia, myelosuppression, gastrointestinal ailments, alopecia are the common adverse effects of paclitaxel treatment. Peripheral neuropathy is the major adverse effect of paclitaxel and increases with increasing cumulative dosage (2). Moreover, Cr El is widely known to cause severe hypersensitivity reactions requiring premedication (4).
It is well recognized that there is inter and intraindividual variability in pharmacokinetic characteristics of paclitaxel. Currently body surface area (BSA) is used in the dosing of paclitaxel, leading to significant interindividual differences in plasma concentrations and risk of severe and treatment limiting adverse effects (5, 6). Quantification of paclitaxel in the blood or plasma samples is necessary to establish pharmacokinetic parameters that can assess inter and intraindividual variability, thereby making necessary dose adjustments to improve therapeutic efficacy and minimizing the adverse effects (7). It is also reported that Cr El has a negative impact on the reproducibility of bioanalysis of paclitaxel due to entrapment of paclitaxel in the micelles of Cr El (8), and various methods are used to nullify the effect of Cr El on quantification of paclitaxel (9).
Therefore, it is necessary to develop a rapid, simple, sensitive, economic and reproducible method to quantify plasma concentrations of paclitaxel in regular clinical practice to promote individualization of treatments.
Till date several methods have been reported for the quantification of paclitaxel in biological matrices using various techniques like high performance liquid chromatography (HPLC), liquid chromatography-tandem mass spectroscopy (LC-MS), micellarelectrokinetic chromatography (MEKC), capillary electrophoresis, immune assays etc (10-12). Most of these methods require tedious, complicated extraction procedures like solid phase extraction (SPE), advanced and expensive instruments like LC-MS which are not available in many hospitals, diagnostic and research labs.
We herewith report on our development of a rapid, simplified, economic and sensitive method of paclitaxel quantification in human plasma by liquid-liquid extraction for application in clinical sample analysis.
METHODS
Instruments and Reagents
The HPLC system - LC- 2010 (Shimadzu Corporation, Kyoto, Japan) liquid chromatographic quaternary auto sampler pump model equipped with UV-Visible detector, Class VP software. C18 HPLC column with 150×4.6mm (i.d) with particle size of 4µm (Waters, Australia), universal guard column with 20×3.9mm i.d and UV detector (Shimadzu), Ultra sonicator (PCI analytics, Vertex enterprises, India), Milli Q water system, Micro centrifuge and vortex shaker.
Paclitaxel (>99% purity) was purchased from Sigma-Aldrich, India. Paclitaxel formulation (Taxeleon 30mg/5mL) was kindly gifted by Neon Laboratories, Mumbai. Acetonitrile, t-butyl methyl ether (t-BME), methanol of HPLC grade, ammonium acetate and formic acid of analytical grade were used. Deionized water purified from Milli-Q water system was used throughout the study. Drug free plasma was obtained from healthy volunteers in our laboratory.
Preparation of stock solution
Stock solution of paclitaxel was prepared by dissolving 10mg of paclitaxel in 10mL of acetonitrile. This stock solution was further diluted for the preparation of working standard solutions with diluent (Acetonitrile and water in 1:1 ratio) to get the concentration range of 6ng/mL to 6µg/mL.
Blood sampling
This method development is a part of pharmacokinetic drug interaction study in cancer patients and has been approved by Institutional ethics committee of our institution (IEC No: IEC-NI/16/Mar/51/16). Blood samples from six cancer patients receiving 260mg of Paclitaxel were collected after procuring signed written informed consent. All the blood samples were collected in heparinized vacutainers. Plasma samples were separated from the whole blood by centrifuging at 3000 rpm for 10min and were stored in refrigerator at -80°C until analysis.
Sample Pretreatment
About 500µL of patient plasma was extracted with 1mL of t-BME twice and vortexed for 1 min. After centrifugation at 2000 rpm for 10 min, the organic layer was transferred into a 10 mL beaker. The t-BME extract was evaporated to dryness, residue was reconstituted with 1mL of diluent and filtered through 0.22 µ pore size nylon membrane filters, about 20 µL of the sample was applied to HPLC analysis as per the proposed chromatographic conditions (13).
HPLC Conditions
Chromatographic separation of paclitaxel was performed using C18 column (150×4.6mm i.d., 4µm particle size, Waters, Australia) with 60% acetonitrile, 40% 10mM ammonium acetate buffer solution and 0.1% formic acid as mobile phase ata flow rate of 1.0 mL/min at ambient temperature. The detection wavelength was 230 nm. Detector output integration was carried using Class VP software to determine peak areas.
Validation
The validation was performed as per ICH Q2(R1) guidelines (14). Calibration curves of paclitaxel were developed using drug free plasma samples spiked with known concentration of paclitaxel standard. The detection limits were calculated from the calibration curve. Intra and inter-day method precision, accuracy, recovery, stability and robustness were assessed using spiked plasma samples with 6-60000 ng/mL paclitaxel standard solution. The recovery was calculated as the difference in the peak area between standard and that spiked to plasma sample.
Statistical methods:
The data was statistically treated using Microsoft excel 2010. Statistical methods like relative standard deviation, percentage and coefficient of determination (r2) were used.
RESULTS
Linearity and detection limits
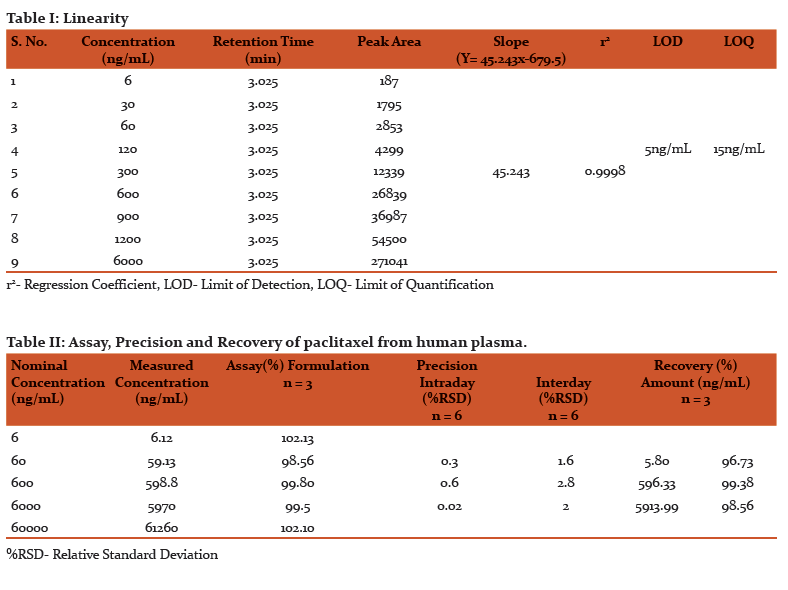
The limit of detection was found to be 5 ng/mL and limit of quantification that can be reliably and reproducibly measured is 15 ng/mL. Linearity of detector response was assessed for extracted plasma standards over the range of 6-6000 ng/mL with an r2 value of 0.9998 (Table I). For 0.5 mL aliquots of plasma samples, linearity was satisfactory between 6 and 6000 ng/mL.
Precision
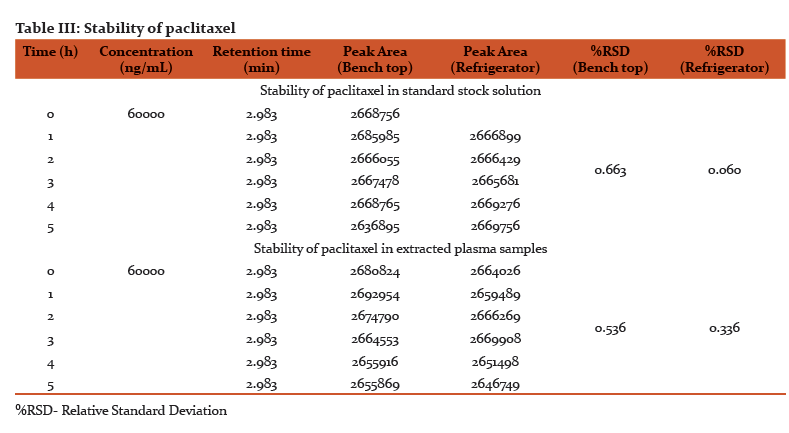
Assay precision was found to be satisfactory in the concentration range of 6-6000 ng/mL. The intraday and interday precision was assessed using six replicates of three concentrations 60, 600, 6000 ng/mL and the relative standard deviation was found to be < 0.7% for intraday and < 3% for interday precision (Table II).
Formulation Assay
Accuracy of the method was established by quantification of paclitaxel in the pharmaceutical dosage form (Taxeleon 30 mg/5mL, Neon laboratories) in the concentration range of 6-60000 ng/mL. The results shows that the peak shape is symmetrical with a good baseline and there was strong correlation between the standard solutions and pharmaceutical dosage form solutions with respect to retention time and peak area. The assay performance data are presented in Table II.
Recovery
Recovery of paclitaxel was estimated by using spiked plasma samples of 500 µL with standard solutions of known concentrations, extraction of plasma samples as per the sample pretreatment procedure, analyzing and comparison of peak areas of paclitaxel standard and extracted plasma samples. Recovery of paclitaxel measured in triplicates at three concentrations (60 ng, 600 ng and 6 µg/mL) was in the range of 96-99% (Table II).
Selectivity and Specificity
Chromatograms of blank plasma, paclitaxel standard in diluent, paclitaxel extracted from patient plasma are illustrated in the Fig 1. The results of extraction from plasma and standard reveal a symmetrical peak shape and good baseline resolution of paclitaxel. Interference due to plasma matrix components was not observed during the analysis. Using this system, the retention time for paclitaxel was 3 min and the runtime for total analysis of each run was efficiently maintained for 5 min.
It is also observed that there is no interference of endogenous peaks, peak shape and retention time being the same for paclitaxel standard in diluent, paclitaxel formulation and extracted plasma samples.
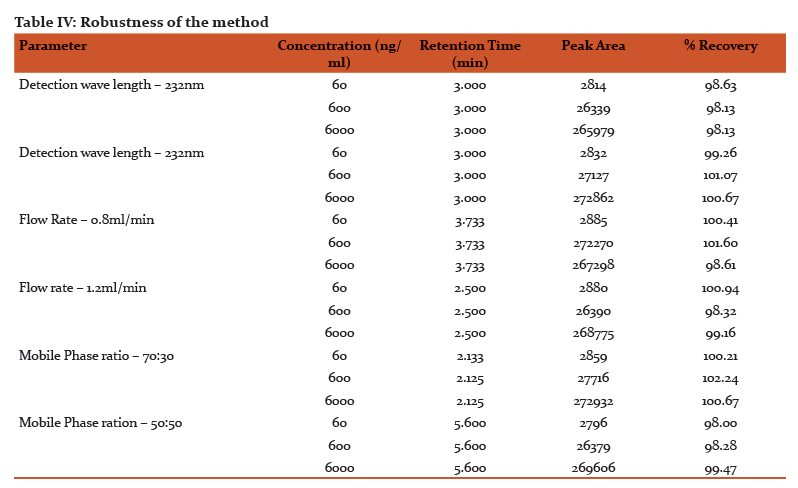
Stability of paclitaxel
The stability of paclitaxel standard solution and extracted plasma were assessed for 6h to determine the optimum requirements for processing and storage during the analysis (Table III). We found that paclitaxel was quite stable in room temperature and at 4°C for the assay duration.
Robustness
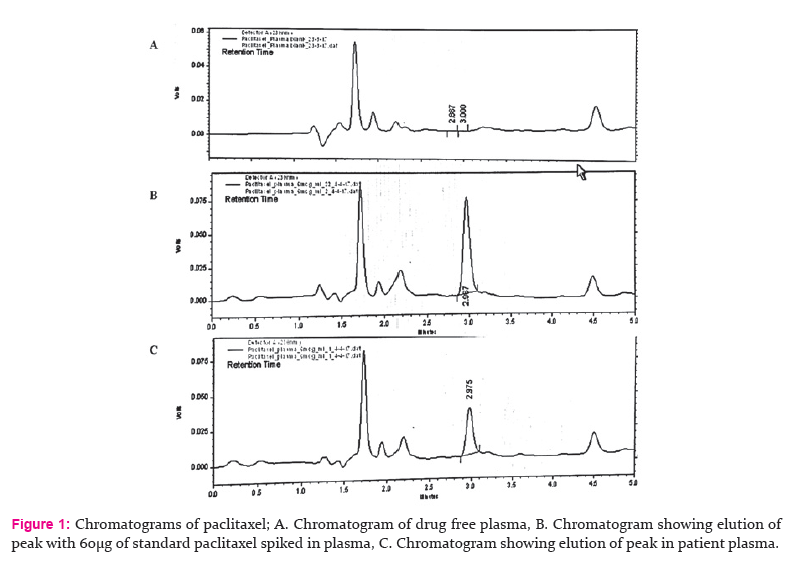
Three chromatographic conditions (Detection wavelength, Flow rate and composition of mobile phase) were altered deliberately at a range of ± 2% and under each condition samples were analyzed to estimate the paclitaxel content. The results reveal that the values were in the acceptance limits of 98-102% with %RSD (Relative Standard Deviation)of less than 2% (Table IV).
Effect of Cremophor El on paclitaxel analysis
It is reported that the bioanalysis of paclitaxel reproducibility is negatively affected by Cr El (9). However, no such effects were observed in our study. This may be due to liquid-liquid extraction with t-BME, that nullifies the ion suppression effects caused by Cr El (15).
DISCUSSION
The limit of detection and limit of quantification of the current developed method is sensitive enough to carry out the clinical studies/pharmacokinetic studies of paclitaxel for 24 h. These are equal to the methods using sophisticated solid phase extraction (10). The value of regression coefficientr2=0.9998 confirmed the linear relationship between the concentration of the drug and area of the peak. This method is precise enough to perform continuous and regular quantification of paclitaxel in human plasma. The minimal variation in %RSD for interday and intraday precision indicates the complete harmony among repeated injection, repeated analysis, intraday and interday study.
The strong correlation between the standard solutions and pharmaceutical dosage form with respect to retention time and peak area establishes the accuracy of the method. The proposed method of analysis was highly selective and specific as peaks of the analyte in spiked plasma and patient plasma were well resolved. Separation of peaks was confirmed by using blank plasma, plasma spiked with analyte and patient plasma containing analyte along with other drugs. The recovery of paclitaxel was efficient and equal to the costly SPE techniques which were used in other methods (10).
The minor deliberate changes made in various experimental parameters did not significantly affect the peak area, recovery of the analyte indicating the robustness of the developed method. The stability of paclitaxel in various conditions has been well studied. In this method stability of paclitaxel in standard solutions and extracted from plasma was assessed for 5 h both at room temp (bench-top) and under refrigeration at 4°C to establish the stability of the analyte during the process of analysis. The %RSD values shows that there were no considerable changes in their concentration after 5 h indicating that samples are stable during analysis.
The extraction solvent used in this method t-BME can eliminate the negative influences caused by the Cr El that are reported in other methods (9). This method was a part of pharmacokinetic drug interaction study of paclitaxel in breast cancer patients. It was successfully applied for quantification of paclitaxel in human plasma and pharmaceutical dosage form. The method is sensitive enough to quantify paclitaxel in the samples upto 24 h of intravenous administration.
CONCLUSION
The current developed RP-HPLC method for quantification of paclitaxel in human plasma is rapid, economic, sensitive and reproducible without negative influence of Cr El. Limit of quantification of this method was 15 ng/mL, making it applicable for clinical pharmacokinetic studies of paclitaxel. A number of methods have been described earlier for quantification of paclitaxel in various biological matrices using various techniques including HPLC. The later involve longer run times, usage of costly SPE procedures and methods to reduce the influence of CrEl. The method described here is equally sensitive yet simpler, using single solvent extraction procedure and having a short retention time of 3 min.
Mobile phase composition, sample extraction using t-BME and the use of C18 (15× 4.6 cm i.d, 4 µm particle size) column have attributed to the sensitivity, precision, reproducibility and shorter retention time of the method, which makes it a very practical method to use.
ACKNOWLEDGEMENTS
Our sincere thanks toNeon laboratories, Mumbai for gifting paclitaxel formulation (Taxeleon 30mg/5mL).
FUNDING
Our sincere thanks to the management, Sri Ramachandra University for supporting the research work in the form of GATE Young Faculty Research Grant for the year 2016-17 (Grant no – 54/Dean/2016)
References:
(1) Huizing MT, Misser VH, Pieters RC, ten Bokkel Huinink WW, Veenhof CH, Vermorken JB, et al. Taxanes: A New Class of Antitumor Agents.Cancer Investigations1995; 13 (4): 381–404.
(2) Jordan MA, Wilson L. Microtubules and Actin Filaments: Dynamic Targets for Cancer Chemotherapy. Current Opinion in Cell Biology 1998; 123–130.
(3) Jordan MA, Wilson L. Microtubules as a Target for Anticancer Drugs. Nature Reviews Cancer 2004; 4 (4): 253–265.
(4) Gligorov J, Lotz JP. Preclinical Pharmacology of the Taxanes: Implications of the Differences. Oncologist 2004; 9 Suppl 2 (suppl 2): 3–8.
(5) Steed H, Sawyer MB. Pharmacology, Pharmacokinetics and Pharmacogenomics of Paclitaxel. Pharmacogenomics2007; 8 (7): 803–815.
(6) Gerritsen-van Schieveen P, Royer B. Level of Evidence for Therapeutic Drug Monitoring of Taxanes. Fundamentals and Clinical Pharmacology2011; 25 (4): 414–424.
(7) Krens SD, McLeod HL, Hertz DL. Pharmacogenetics, Enzyme Probes and Therapeutic Drug Monitoring as Potential Tools for Individualizing Taxane Therapy. Pharmacogenomics 2013; 14 (5): 555–574.
(8) Sparreboom A, Zuylen L Van, Brouwer E, Loos WJ, Bruijn P De, Gelderblom H et al. Cremophor EL-Mediated Alteration of Paclitaxel Distribution in Human Blood?: Clinical Pharmacokinetic Implications.Cancer Research 1999; 1454–1457.
(9) Andersen A, Warren DJ, Brunsvig PF, Aamdal S, Kristensen GB, Olsen H. High Sensitivity Assays for Docetaxel and Paclitaxel in Plasma Using Solid-Phase Extraction and High-Performance Liquid Chromatography with UV Detection. BMC Clinical Pharmacology 2006; 6 (1): 2.
(10)HendrikxJJMA, Rosing H, SchinkelAH, Schellens JHM, Beijnen JH. Quantification of Taxanes in Biological Matrices: A Review of Bioanalytical Assays and Recommendations for Development of New Assays. Bioanalysis 2014; 6 (7): 993–1010.
(11) Palmas RA, Monteiro J, Fresco P. Analytical Methods for Taxanes Quantification in Diluted Formulations and Biological Samples and Their Applications in Clinical Practice. International Journal of Pharmacy and Pharmaceutical Sciences 2014; 6 (6): 17–23.
(12) Khan I, Iqbal Z, Khan A, Hassan M, Nasir F, Raza A et al. A simple, rapid and sensitive RP-HPLC-UV method for the simultaneous determination of sorafenib and paclitaxel in plasma and pharmaceutical dosage forms: Application to pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci., 2016;DOI: 10.1016/j.jchromb.2016.08.029.
(13) Yonemoto H, Ogino S, Nakashima MN, Wada M, Nakashima K. Determination of Paclitaxel in Human and Rat Blood Samples after Administration of Low Dose Paclitaxel by HPLC-UV Detection. Biomedical Chromatography 2007; 21: 310-317.
(14)https://www.gmp-compliance.org/guidelines/gmp-guideline/ich-q2r1-validation-of-analytical-procedures-text-and-methodology (Last accessed: October 28, 2017)
(15) Bhaskar V. Identification and Reduction of Matrix Effects Caused by Cremophor EL in Bioanalysis Using Liquid Chromatography/Tandem Mass Spectrometry. Analytical and Bioanalytical Techniques 2013; 4 (3): 167.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





